
 English
English
 French
French
 Pubmed
Pubmed Google Scholar
Google Scholar Cross Ref
Cross Ref Visitor 725689
Visitor 725689Physiopathologie de la bronchopulmonaire chronique obstructive - BPCO
Physiopathology of chronic obstructive pulmonary disease - COPD
C. Préfaut
Département de Physiologie Clinique
CHRU de Montpellier, INSERM 1046 – FRANCE
Corresponding author
Pr. Christian PREFAUT
Département de Physiologie Clinique. CHRU de Montpellier,INSERM 1046
E-mail: c-prefaut@chu-montpellier.fr
ABSTRACT
Chronic obstructive pulmonary disease (COPD) is defined as a progressive onset disease that may be treated and prevented. COPD is characterized by flow limitation and incomplete reversibility that is the consequence of an inflammatory response related to harmful particles, most commonly tobacco, and usually has a systemic impact.
COPD is first a disease of a single-organ: the broncho–pulmonary system, which affects mainly the small airways, resulting bronchial obstruction. The latter causes static hyperinflation and distension that worsens during exercise (dynamic hyperinflation) and induces the essential clinical signs of the disease: exertional dyspnea. COPD manifests into a general or systemic disease, characterized mainly by associated co-morbidities, such as muscular atrophy. Muscular atrophy worsens the prognosis of the patient and worsens dyspnea (muscular dyspnea). This atrophy is the result of patient’s sedentary lifestyle, which must be strongly advised against. It appears a multi-factorial myopathy during which oxidative stress plays an important role; all coming together as the muscle self-destruct and thus are incapable to regenerate. The treatment of COPD should aim to include the two components of this disease: broncho-dilatators for the single-organ disease and physical rehabilitation for the systemic disease.
KEYWORDS: COPD, tobacco, bronchial obstruction, hyperinflation, muscular atrophy, rehabilitation.
RÉSUMÉ
La broncho pneumopathie obstructive chronique (BPCO) a défini comme une maladie d’apparition progressive que l’on peut traiter et prévenir, qui est caractérisée par une limitation des débits, non complètement réversible, qui est la conséquence d’ une réponse inflammatoire liée aux particules nocives, en particulier le tabac, et qui s’accompagne d’un retentissement systémique.
La BPCO est d’abord une maladie d’organe: l’appareil broncho-pulmonaire, qui atteint essentiellement les petites voies aériennes. Ceci se traduit par une obstruction bronchique. Cette dernière va entrainer une hyper inflation statique ou distension qui s’ aggrave au cours de l’effort (hyperinflation dynamique) et induit le signe clinique essentiel de la maladie: la dyspnée d’effort.
La BPCO est ensuite une maladie générale ou systémique essentiellement caractérisée par des comorbidités. L’une d’entre elles, l’atrophie musculaire conditionne le pronostic des patients et intervient comme facteur d’aggravation de la dyspnée (dyspnée musculaire). Cette atrophie est en premier lieu la conséquence de la sédentarisation des patients contre laquelle il faut absolument lutter. Puis apparait une myopathie multi factorielle au cours de laquelle le stress oxydant joue un rôle important, tout se passant comme si le muscle s’autodétruisait et était incapable de se régénérer. Le traitement de la BPCO doit s’attaquer aux deux versants de la BPCO. Les broncho-dilatateurs pour la maladie d’organe, la réhabilitation respiratoire pour la maladie générale.
MOTS CLES: BPCO, tabac, obstruction bronchique, hyperinflation, atrophie musculaire, réhabilitation.
INTRODUCTION
Un task force commun de l’American Thoracique Society et de l’European Respiratory Society a défini en 2004 la broncho pneumopathie obstructive chronique (BPCO) comme une maladie d’apparition progressive que l’on peut traiter et prévenir, qui est caractérisée par une limitation des débits, non complètement réversible, qui est la conséquence d’ une réponse inflammatoire liée aux particules nocives, en particulier le tabac, et qui s’accompagne d’un retentissement systémique [1]. Cette définition a été reprise par GOLD en 2008. C’est dire, si elle a mis longtemps à se dessiner, qu’elle est maintenant totalement consensuelle.
De plus, elle offre à ce texte son plan. En effet elle dégage les deux caractéristiques essentielles de la BPCO. C’est d’abord une maladie d’organe ou primaire: le système broncho-pulmonaire mais c’est ensuite une maladie systémique ou générale. Nous entrons là dans la définition de toute maladie chronique: maladie générale à point de départ organique. La BPCO est donc une maladie générale à point de départ respiratoire.
Nous envisageron ssuccessivement la physiopathologie de la BPCO maladie primaire respiratoire puis la BPCO maladie systémique en insistant sur les atteintes musculaires qui font essentiellement le pronostic de cette affection.
PHYSIOPATHOLOGIE DE LA BPCO MALADIE RESPIRATOIRE
Les facteurs de risque
En premier lieu vient le tabac dont on considère qu’il est la cause de 80 à 90% des BPCO. Parmi ses nombreux constituants insistons sur le rôle de certains radicaux libres de l’oxygène entrainant l’ apparition d’un stress oxydant et d’une inflammation ici au niveau bronchique mais que nous retrouverons à tous les étages de la maladie générale.
Vient ensuite la combustion de la biomasse et ce en particulier dans les pays en voie de développement, surtout lorsque l’aération de la pièce ne permet pas une évacuation satisfaisante de la fumée et autres produits de la combustion.
Les jouent également un rôle, les professions les plus susceptibles étant les mineurs, les travailleurs du béton et de manière plus générale toutes celles qui font inhaler des poussières minérales organiques ou chimiques.
La susceptibilité génétique est également un facteur de risque et ce, malgré sa complexité d’analyse, pour 2 raisons majeures. Il est clair que le déficit en alpha-1 antitrypsine entraine l’apparition d’un emphysème pan lobulaire, mais cela ne concerne que 2% des BPCO. Par ailleurs et surtout, seul 1/3 des fumeurs présente, à tabagisme égal, une BPCO. Le fait associé qu’il existe des familles de BPCO laisse supposer l’existence d’un facteur génétique ou épi-génétique. On a actuellement mis en évidence des gènes candidats essentiellement sur les chromosomes 4 et 5.
Les données anatomiques
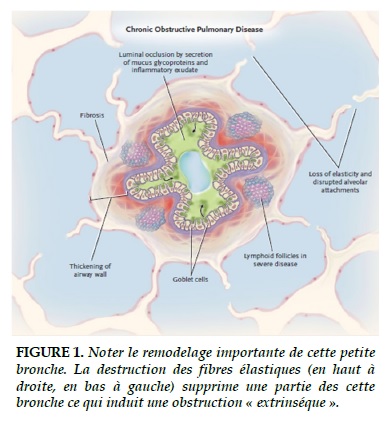
La BPCO est d’abord une maladie des petites voies aériennes (dont le diamètre intérieur est < à 2mm, soit à partir de la 6ème à la 8ème génération bronchique selon les sujets), ce qui la différencie de la bronchite chronique maladie des grosses voies aériennes. L’inhalation d’irritants produit une réponse inflammatoire (neutrophiles, lymphocytes T CD4…) aboutissant à un remodelage bronchique caractérisé par un épaississement de la paroi, une métaplasie des glandes caliciformes, une diminution des cellules ciliées, une hypertrophie des muscles lisses [2, 3] (Figue 1).
L’ensemble de ces données va induire une obstruction aggravée par la sécrétion de mucus et qualifiée d’intrinsèque. Comme nous le verrons ci-dessous, cette obstruction va entrainer une diminution des débits aériens à travers le système bronchique, c’est donc une obstruction « débitmétrique ».
Le parenchyme pulmonaire est également atteint par des lésions de type emphysémateux qui détruisent les alvéoles (et les capillaires). Les alvéoles contiennent des fibres élastiques dont l’ensemble, que l’on peut imaginer comme un continuum, sert d’attache, de hauban aux petites bronches. Ces dernières qui n’ont aucun squelette, ne serait-ce que cartilagineux, sont donc maintenues ouvertes par ce réseau élastique. La destruction alvéolaire fait disparaitre ce support élastique et les petites bronches ont tendance à se fermer [2, 3] (Figue 1) ce qui contribue à leur obstruction, ce dernier type étant qualifié d’extrinsèque et dans l’état actuel de nos connaissances totalement irréversible.
Cette perte des fibres élastiques a pour autre conséquence une diminution de la force de rétraction élastique du poumon. Ici nous devons faire un rappel de physiologie respiratoire. La fin de toutes les expirations spontanées d’un sujet se situe à un niveau pulmonaire précis, appelé niveau ventilatoire de repos (délimitant la capacité résiduelle fonctionnelle ou CRF), qui résulte de l’équilibre entre les forces opposées de rétraction élastique de la cage thoracique et du poumon. Si la force de rétraction élastique du poumon diminue un nouvel équilibre doit être trouvé. Il se trouve obligatoirement à un plus grand volume pulmonaire. Il y a donc nécessairement une plus grande capacité résiduelle fonctionnelle c’est ce que l’on appelle l’hyperinflation alvéolaire statique ou plus communément la distension pulmonaire. Elle est irréversible et liée l’obstruction extrinsèque que l’on peut qualifier maintenant de « volumétrique » puisqu’en relation avec les volumes pulmonaires.
Les conséquences fonctionnelles
L’obstruction est évidemment la première conséquence objective de cette maladie des petites voies aériennes qui sera difficile à mettre en évidence parce que située dans la zone « silencieuse » du poumon, c’està-dire celle qui ne « parle » pas, ou mal, du point de vue de l’exploration fonctionnelle, et de celui de la clinique. Comme nous le verrons ci-dessous c’est l’obstruction volumétrique qui parlera en premier mais le diagnostic ne sera formel que lorsque l’ obstruction débitmétrique apparaitra.
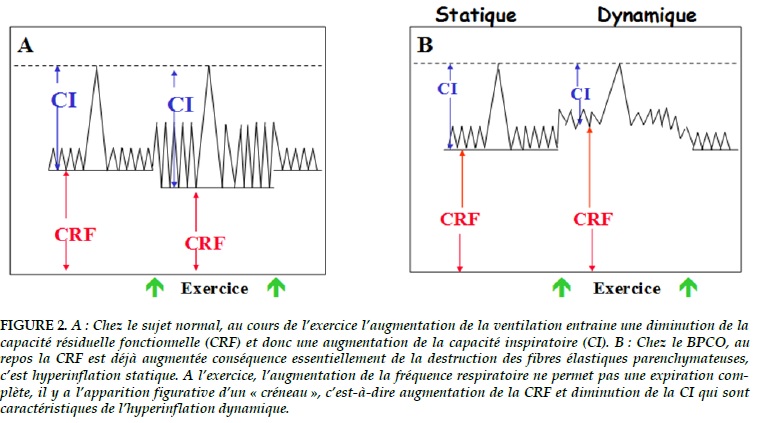
La dyspnée d’effort est en fait le premier signe clinique ressenti par le patient. Comment s’explique-t-elle ? Au repos l’obstruction va essentiellement ralentir l’expiration, mais comme il n’y a pas de limite de temps celle-ci sera complète. A l’exercice, physiologiquement, la fréquence respiratoire s’accélère pour augmenter l’apport en oxygène. Le temps nécessaire à l’expiration n’est plus suffisant, celle-ci est donc incomplète. Le volume expiratoire est ainsi diminué et le niveau moyen de fin d’expiration, donc la CRF, est très supérieur niveau ventilatoire de repos, c’est le signe du créneau que nous observons sur la Figure 2. La capacité pulmonaire totale (CPT) est la somme de la CRF et de la capacité inspiratoire (CI). Puisqu’au cours de l’exercice, ici à charge constante, la CPT reste inchangée, l’augmentation de la CRF entraine une diminution de la CI. Ce sont les 2 signes caractéristiques de l’hyperinflation dynamique (HD).
Cette HD a plusieurs conséquences. A titre d’exemple, elle va provoquer rapidement une diminution du volume courant et donc une respiration de type « petit volume courant, fréquence respiratoire élevée », très défavorable pour les échanges respiratoires. Par ailleurs cette ventilation à haut volume pulmonaire place les muscles respiratoires dans une position géométrique défavorable induisant une fatigue. L’ensemble de tous les phénomènes induits par l’HD explique la dyspnée d’effort.
La dyspnée à l’exercice est un excellent modèle pour mettre en évidence l’hyperinflation dynamique, mais ce qui a provoqué son apparition c’est l’hyperventilation. En d’autres termes tout facteur d’hyperventilation entrainera une HD et donc une dyspnée, par exemple une exacerbation. En fin d’évolution de la maladie lorsque l’obstruction est sévère et le déconditionnement (voir ci-dessous) maximal, le patient hyperventile au repos il y a alors apparition d’une hyperinflation dynamique est d’une dyspnée de repos. Il y a longtemps que l’on sait que l’ hyperinflation dynamique explique 45% de la variance de la dyspnée ce qui est considérable [4].
Les troubles des échanges gazeux, sont la conséquence des différentes anomalies du rapport entre la ventilation et la perfusion pulmonaire. L’effet shunt, le plus banal, représente les espaces non ventilés mais perfusés. Le sang qui passe en regard de ces zones n’est pas oxygéné, il y a donc apparition d’une hypoxie qui ne peut être compensée. L’effet espace mort représente quant à lui les espaces ventilés mais non perfusés, le CO2 ne peut donc être éliminé. Cette altération est compensable au début par une hyperventilation, en fin d’évolution de la maladie si cette compensation est insuffisante il y a apparition d’une hypercapnie.
Le retentissement de la BPCO sur la circulation pulmonaire a, au moins, une double origine. Tout d’ abord l’hyperinflation statique, ou distension, comprime les capillaires pulmonaires par ailleurs l’ hypoxie alvéolaire induit, pour favoriser les échanges gazeux dans les zones saines, une vasoconstriction hypoxique. L’ensemble de ces deux phénomène sera facteur d’hypertension artérielle pulmonaire et à terme d‘insuffisance ventriculaire droite.
Rapportons enfin les troubles de la capacité de transfert de l’oxygène. Ils sont la conséquence, d’une part, des altérations du rapport ventilation/perfusion décrits ci-dessus. Par ailleurs, dans les zones purement emphysémateuses il y a disparition parallèle de la ventilation et de la perfusion, diminution de la surface d’échange donc absence totale d’échange de l’oxygène (et du CO2). Ceci est globalement sans conséquence au repos mais entraine l’apparition d’ une hypoxémie et d’une désaturation oxyhémoglobinée à l’exercice. En effet l’exercice induit une augmentation du débit cardiaque, le sang passe trop vite sur une surface d’échange trop étroite et ne peut s’oxygéner correctement.
Le diagnostic et l’évaluation fonctionnelle de la BPCO
La suspicion clinique du diagnostic de la BPCO est difficile à faire. Le seul signe clinique réel de la maladie est la dyspnée d’effort, mais elle est délicate à retrouver car le patient s’y adapte physiologiquement et surtout environnementallement: ne monte plus d’ escalier, se déplace de manière motorisée… La toux est un phénomène banal non discriminant et l’expectoration est peu importante les sécrétions étant bloquées au niveau des petites voies aériennes.
Le diagnostic doit donc être évoqué sur les facteurs de risque: sujet de plus de 40 ans ayant un tabagisme d’au moins 10/15 paquets-année. Mais aussi chez tous sujets consultant trop fréquemment pour une surinfection pulmonaire.
Le diagnostic positif de la BPCO est fonctionnel, il faut donc réaliser une spirographie. Lorsque, 1) le rapport Volume Expiratoire Maximum en 1 seconde sur Capacité Vitale (en général forcée) (VEMS/CVF) est inférieur [a] à 70% sur un appareil ne donnant pas les valeurs normales, ou [b] à la limite inférieure de la normale pour les autres ; 2) que ces données séparées ne sont pas complètement réversibles sous broncho-dilatateur, le diagnostic de BPCO est confirmé.
Il est intéressant de commenter ces données. Le diagnostic de BPCO n’est fait qu’à partir du rapport VEMS/CV, celui-ci dépendant essentiellement de la diminution du VEMS (donc des débits bronchiques), la CVF étant normale ou diminuée chez le BPCO. Il met donc en évidence une obstruction débitmétrique. Le problème est que le VEMS est essentiellement le témoin d’une obstruction prédominant sur les grosses voies aériennes ou généralisée à l’ensemble des bronches. Ce paramètre est donc relativement peu sensible mais c’est le seul reconnu par les recommandations internationales.
En pratique il n’est pas rare d’observer d’abord une obstruction volumétrique c’est-à-dire une augmentation isolée de la CRF (hyperinflation statique). La CRF n’est pas toujours mesurée, mais elle s’accompagne souvent d’une diminution de la Capacité Vitale Forcée (CVF), donnée par tous les spirographes simples. Ainsi si la mise en évidence d’une diminution isolée de la CVF chez un sujet à risque de BPCO ne permet pas le diagnostic de cette maladie, elle permet au moins de le suspecter et surtout de lui recommander fortement d’arrêter son tabagisme.
Il est utile de mesurer les gaz de sang à la recherche d’une hypoxémie voire d’une hypercapnie. Il est plus facile de suivre la saturation oxyhémoglobinée du patient par l’intermédiaire d’un oxymètre de pouls. Celle-ci reste longtemps normale, malgré une hypoxémie modérée, conséquence physiologique la forme de la courbe de fixation de l’oxygène sur l’ hémoglobine. En clair la saturation rend bien compte de l’état d’oxygénation des patients. Puis la désaturation apparait et s’accroit signe d’une évolution de la maladie. Lorsqu’elle devient inférieure à 90%, la désaturation est très sévère et l’état de patient nécessite sa mise sous oxygène au moins 15 heures par jour.
On pourrait mesurer la capacité de transfert ou diffusion de l’oxyde de carbone, qui pourrait se montrer très abaissée révélant l’existence de lésions emphysémateuses franches. Cet examen a en fait un intérêt essentiellement physiopathologique.
Par contre l’évaluation de la tolérance à l’effort est de grand intérêt. Sa diminution signe de manière schématique le passage de la maladie primaire à la maladie chronique, de la maladie d’organe à la maladie générale. Il n’est pas nécessaire de réaliser une épreuve d’effort, un simple test de marche de 6 minutes (TDM6) est suffisant pour mettre en évidence une intolérance à l’exercice.
PHYSIOPATHOLOGIE DE LA BPCO: MALADIE SYSTEMIQUE, D’ABORD MUSCULAIRE
Les atteintes systémiques font parties de la définition de la BPCO et plus largement sont inhérentes à toute pathologie chronique. D’un point de vue sémantique il est nécessaire de différencier les conséquences systémiques de la maladie et ses comorbidités. Stricto sensu une atteinte systémique implique un lien de causalité, par exemple l’hyperinflation statique comprime les capillaires pulmonaires ce qui induit une hypertension artérielle pulmonaire qui à son tour est facteur d’insuffisance cardiaque droite. Les comorbidités, comme leur nom l’indique, sont des atteintes organiques associées, si l’on reste dans l’ appareil cardio-vasculaire l’exemple est celui des cardiopathies ischémiques.
La réalité est sans doute plus complexe puisque les facteurs de risque comme le tabagisme ou la sédentarité sont les mêmes, nous considèrerons donc qu’il s’agit de l’évolution de la maladie et utiliserons indifféremment les 2 termes. D’un point de vue pratique il faut savoir qu’au moment où l’on fait le diagnostic de BPCO, un malade sur deux présente déjà une comorbidité. Ce qui importe médicalement c’est de prévenir ces différentes manifestations de la maladie chronique ou tout au moins de les diagnostiquer le plus rapidement possible pour ralentir leur évolution.
La BPCO et ses « comorbidités »
L’ensemble des organes est touché [5, 6], nous reparlerons de l’atteinte musculaire dont la prévalence est à nos yeux la plus importante, mais masquée par différentes appellations. C’est ainsi que l’on parle à priori indifféremment de « dénutrition », mais qui devrait être corrigée par une renutrition ce qui n’est pas le cas, de cachexie bien réelle mais qui n’arrive qu’en fin d’évolution et qui n’est que l’aggravation de l’atrophie musculaire le terme que l’on devrait utiliser.
Les complications cardio–vasculaires sont les plus graves puisque dans l’étude TORCH [7] elles représentent 27% de la mortalité des patients. Le cancer broncho-pulmonaire mérite une mention particulière non pas tant par sa fréquence variable selon les auteurs (3 à 18%) mais par le fait qu’il passe souvent inaperçu chez ces patients respiratoires, ce qui n’ exclut pas l’apparition d’autres types de cancers (21% de mortalité pour l’ensemble des cancers dans TORCH [7]). L’ostéoporose accompagne habituellement l’atrophie musculaire. L’anémie est courante dans toutes les maladies chroniques et doit être recherchée. Le surpoids et l’obésité sont de plus en plus fréquents dans la BPCO, au moins 1/3 des patients en France, ce qui aggrave la dyspnée et la sédentarité des patients et induit syndromes métaboliques et de diabètes de type II. Le syndrome d’ apnées obstructives du sommeil est associé à la BPCO (overlap syndrom) dans 10 à 15% des cas. Signalons enfin la grande prévalence de l’anxiété (50 75%) et de la dépression (30 - 50%) chez nos patients [5, 6].
Les mécanismes physiopathologiques proposés sont nombreux tant il est difficile d’établir des liens de causalités. Une théorie fait intervenir comme mécanisme central [5] l’inflammation de bas grade. C’est effectivement une constante dans les maladies chronique, dans la BPCO il est suggéré que l’inflammation bronchique induite par le tabac, induirait une inflammation systémique (il faut comprendre sanguine) qui elle-même induirait l’inflammation des autres organes. L’hypothèse est simpliste, pas forcément fausse mais surement insuffisante pour expliquer la complexité des phénomènes. Aux côtés de l’inflammation de bas grade il est proposé d’autres mécanismes: le stress oxydant (généralement lié à cette dernière), la sédentarité, l’hypoxémie, la génétique, le vieillissement…
L’altération musculaire de la BPCO et ses conséquences
L’altération des muscles squelettiques du BPCO est connue depuis les années 80 mais sa confirmation expérimentale est plus récente fin des années 90 et début des années 2000 [8].
Il a d’abord été montré dès 1998, par des scanners de cuisse, une diminution d’un tiers de la surface de section (atrophie musculaire) de celle-ci et parallèlement une réduction de moitié de la capacité d’endurance du quadriceps [9].
Ces travaux initiaux sur la dysfonction musculaire du BPCO ont largement été confirmés par la suite. On considère actuellement que la diminution de la force musculaire du quadriceps, liée physiologiquement à sa surface de section, est d’un tiers. La réduction de l’endurance de ce même muscle, facteur physiologique essentiel car étroitement lié aux activités de la vie quotidienne, est de 60% [8]. Le plus impressionnant est que cette atrophie n’est pas la conséquence d’une altération typologique homogène du muscle.
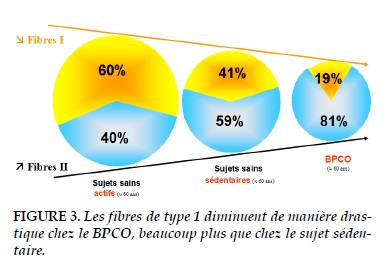
Rappelons, d’une manière schématique, que le muscle est essentiellement constitué de fibres de types I, oxydatives, rouges, lentes, résistantes à la fatigue et de fibres de type II, glycolytiques, blanches, rapides, fatigables et produisant de l’acide lactique. Tous les travaux montrent (Figure 3 ; [8]) que les fibres de type I diminuent de manière drastique chez le sujet BPCO, alors que les fibres de type II augmentent proportionnellement. Cette disparition des fibres de type I a probablement une double origine, d’une part destruction de certaines d’entre elles mais également transformation en fibres de type II. Cette altération typologique s’accompagne d’une réduction de la capillarisation et d’une perte des enzymes de la voie aérobie.
Ainsi le quadriceps muscle de type endurant par excellence a perdu son caractère oxydatif chez le patient BPCO et s’est transformé en muscle fatigable. Cette dégradation typologique atteint à des degrés variables les différents groupes musculaires.
Les muscles des membres supérieurs par exemple ne sont atteints que beaucoup plus tardivement, en fin d’évolution de la maladie.
Le diaphragme est un cas particulier puisque celui du BPCO possède plus de fibres de type I que chez un sujet normal. C’est ce que l’on observe lorsqu’un muscle est entrainé. Or le seul muscle bénéficiant d’un entrainement constant chez le BPCO est bien le diaphragme puisqu’il doit lutter en permanence contre le frein que représente l’obstruction bronchique.
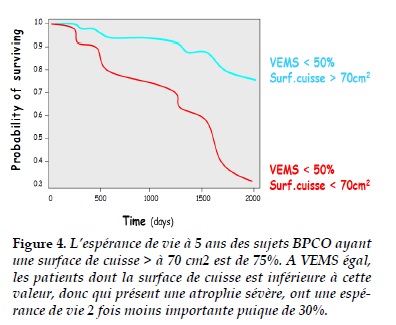
Les conséquences de ces altérations anatomiques, histologiques et fonctionnelles des muscles squelettiques du BPCO sont majeures. Du point de vue de la mortalité, l’espérance de vie est 2 fois moins importante, à VEMS égal, si l’atrophie du quadriceps est sévère (Figure 4 [10]).
En d’autres termes ce qui fait le diagnostic de la maladie c’est l’obstruction mais ce qui fait son pronostic c’est l’atrophie du quadriceps. Concernant la morbidité les conséquences de l’altération de la typologie musculaire sur les activités de la vie quotidienne (et donc la qualité de vie) sont tout aussi sévères. Lors d’une simple déambulation, les fibres oxydatives de type I sont insuffisantes pour générer l’énergie nécessaire.
Le relai est alors pris pas les fibres glycolytiques de type II, ce qui aboutit à une production d’acide lactique. Qu’il soit ou non tamponné en CO2, celui-ci va stimuler les métabo-récepteurs musculaires, voire directement les centres nerveux respiratoire pour provoquer une hyperventilation destinée à éliminer l’excès d’acidité (alcalose ventilatoire), mais celle-ci va évidemment augmenter la dyspnée du patient, c’est ce que l’on appelle le « cercle vicieux de la dyspnée ».
La notion importante est que la dyspnée du patient du BPCO est multi factorielle. A côté de la dyspnée d’origine respiratoire, dont nous avons vu qu’elle était essentiellement due l’hyperinflation dynamique qui expliquait 45% du phénomène, il faut donc insister sur la dyspnée musculaire que nous venons de décrire. Sa part est à peine inférieure (+/- 40%) à la précédente dans l’explication de la globalité de la dyspnée. Il est intéressant de noter que l’on retrouve chez l’insuffisant cardiaque chronique les mêmes anomalies musculaires que chez le BPCO. La dyspnée qu’il présente est de manière prépondérante d’origine musculaire et beaucoup plus discrètement respiratoire. Les causes purement cardiaques d’une dyspnée chronique, n’ont pas fait leur preuve.
Les causes de l’altération musculaire de la BPCO
La cause la plus anciennement connue d’altération musculaire est l’inactivité physique. Il faut rappeler que physiologiquement la masse musculaire est d’ abord régulée par l’activité physique. Le phénomène est tellement important qu’une immobilisation de 3 semaines induit une atrophie musculaire et une perte de 20% des fibres de type I, c’est ce que l’on appelle le déconditionnement. Chez les sujets BPCO la réduction d’activité physique est la conséquence d’ une sédentarisation [11], à titre d’exemple le temps de marche et la station debout chez les BPCO sont diminués d’un tiers par rapport aux sujets normaux. La dysfonction musculaire est corrélée à cette sédentarité [9, 10].
Un cercle vicieux va progressivement s’installer. La sédentarité chronique du BPCO provoque le déconditionnement, c’est-à-dire la perte des fibres de type I ci-dessus décrite. Lors d’un exercice il y a apparition de la dyspnée musculaire qui va aggraver la dyspnée respiratoire préexistante. La malade ressentant cette aggravation de la dyspnée à l’exercice va, logiquement, encore diminuer son activité physique, ce qui va accélérer la disparition des fibres de type I et ainsi de suite.
Un cercle vicieux s’est donc installé dit du déconditionnement, il est totalement superposable au cercle vicieux de la dyspnée décrit précédemment et les 2 termes sont utilisés indifféremment pour décrire l’aggravation de la dyspnée d’effort du patient en conséquence de l’accentuation de sa sédentarité. Le but de la réhabilitation respiratoire est de renverser ce cercle vicieux en réintroduisant l’activité physique dans la vie du patient, de façon à diminuer sa dyspnée musculaire et donc d’améliorer ses activités de la vie quotidienne c’est à dire sa qualité de vie.
Si l’on se reporte à la figure 3 [8], on constate qu’un sujet sédentaire sains possède 1/3 de fibres de type I de moins qu’un sujet sain non sédentaire, c’est-à-dire pratiquant au moins 2h30 min d’activité physique par semaine. C’est ce que nous avions décrit. L’observation surprenante est qu’un patient BPCO sédentaire a perdu 1/3 supplémentaire de fibres de type I. Bien sûr on peut supposer que la sédentarisation est plus ancienne, ce qui n’est pas démontré, ou plus importante chez les patients, mais la différence est trop flagrante pour être passée sous silence. Elle suggère l’existence d’autres facteurs qui a côté de la sédentarité pourraient expliquer l’altération des muscles du patient BPCO. C’est la notion de myopathie acquise chez ces patients [8].
L’origine de cette myopathie est multi factorielle. On pense d’abord à une étiologie iatrogène avec les corticoïdes prescrits lors des exacerbations. Mais lorsque ce type de sujet est exclu des études on retrouve les mêmes altérations. Plusieurs éléments doivent être alors considérés comme l’inflammation et le stress oxydant systémiques bien sûr mais surtout locaux. Comme déjà signalés ces deux éléments se retrouvent au niveau sanguin. Au niveau musculaire on les retrouve également au repos. Par contre lors d’un exercice du quadriceps seul le stress oxydant (SO) s’aggrave, quel que soit le marqueur utilisé [12]. Cet accroissement du SO qui est corrélée à la dysfonction musculaire et aggravé par l’hypoxie mais partiellement corrigée par une cure d’antioxydants [13].
Ainsi le stress oxydant joue un rôle important dans la myopathie du BPCO, ce qui n’exclut pas évidemment l’existence d’autres phénomènes. Il est important de signaler que chaque exacerbation aggrave la maladie musculaire et donc le pronostic de la maladie. Ceci n’est guère étonnant puisque les exacerbations s’accompagnent d’une inflammation et d’un stress oxydant majeur sans oublier la effets potentiellement délétère d’un traitement par corticoïde. Il est intéressant de noter qu’une myopathie acquise de même nature a été décrite chez les insuffisants cardiaques chroniques.
La question qui se pose est de savoir quels sont les mécanismes cellulaires à la base des altérations musculaires. Physiologiquement le maintien de la masse musculaire, renouvelée en permanence, dépend d’un équilibre précis entre dégradation et synthèse des protéines. Schématiquement la dégradation dépend essentiellement du système ubiquitine-proteasome, stimulé par le stress oxydant et par l’inflammation, alors que la synthèse dépend des facteurs de croissance comme IGF1, VEGF… Au cours de la BPCO in semble que les 2 phénomènes soient perturbés.
Les données les plus importantes concernent le complexe ubiquitine-proteasome qui est sur surexprimé dans cette pathologie, facilitant ainsi la dégradation musculaire [14]. Les voies thérapeutiques pourraient être dans l’immédiat des antioxydants mais spécifiques des phénomènes observés dans le quadriceps du BPCO. Dans l’avenir les inhibiteurs du complexe ubiquitine-proteasome pourraient se révéler utiles.
CONCLUSION
La BPCO est d’abord une maladie d’organe: l’appareil broncho-pulmonaire, qui atteint essentiellement les petites voies aériennes. Ceci se traduit par une obstruction bronchique. Cette dernière va entrainer une hyper inflation statique ou distension qui s’ aggrave au cours de l’effort (hyperinflation dynamique) et induit le signe clinique essentiel de la maladie: la dyspnée d’effort.
La BPCO est ensuite une maladie générale ou systémique essentiellement caractérisée par des comorbidités. L’une d’entre elles, l’atrophie musculaire conditionne le pronostic des patients et intervient comme facteur d’aggravation de la dyspnée (dyspnée musculaire). Cette atrophie est en premier lieu la conséquence de la sédentarisation des patients contre laquelle il faut absolument lutter. Puis apparait une myopathie multi factorielle au cours de laquelle le stress oxydant joue un rôle important, tout se passant comme si le muscle s’ autodétruisait et était incapable de se régénérer. Le traitement de la BPCO doit s’attaquer aux deux versants de la BPCO. Les broncho-dilatateurs pour la maladie d’organe, la réhabilitation respiratoire pour la maladie générale.
CONFLIT D’INTERETS
Aucun.
REFERENCES
1. Celli BR, MacNee W. Standards for the diagnosis and treatment of patients with COPD a summary of the ATS/ERS position paper. Eur Respir J 2004; 23: 932.
2. Barnes PJ. Small airways in COPD. N Engl J Med 2004; 350: 26.
3. Hogg JC, Chu F, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med 2004; 350: 2645.
4. O'Donnell DE, Webb KA. Exertional breathlessness in patients with chronic airflow limitation. The role of lung hyperinflation. Am Rev Respir Dis 1993; 52: 70.
5. Barnes PJ, Celli BR. Systemic manifestations and comorbidities of COPD. Eur Respir J 2009; 33: 1165.
6. Agusti A, Calverley PM, Celli B et al. Characterisation of COPD heterogeneity in the ELIPSE cohort. Respir Res 2010; 11: 122.
7. McGarvey LP, Anderson JA et al. Ascertainment of cause-specific mortality in COPD: operations of the TORCH clinical endpoint committee. Thorax 2007; 62: 411.
8. Couillard A, Préfaut C. From muscle disuse to myopathy in COPD: potential contribution of oxydative stress. Eur Respir J 2005; 26: 703.
9. Serres I, Gautier V., Varray A., Préfaut C. Impaired squeletal muscle endurance related to physical inactivity and altered lung function in COPD patients. Chest 1998; 113: 900.
10. Marquis K, Jobin J, Maltais F et al. Mid thigh muscle cross-sectional area is a better predictor of mortality than body mass index in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2002; 166: 809.
11. Pitta F., Troosters T., Decramer M. et al. Characteristics of physical activities in daily life in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2005; 171: 972.
12. Couillard A, Maltais F, Préfaut C et al. Exerciseinduced quadriceps oxydative stress and peripheral muscle dysfonction in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2003; 167: 1664.
13. Koechlin C, Couillard A, Préfaut C et al. Does oxydative stress alter quadriceps endurance in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2004; 169: 1022.
14. Caron MA, Debigaré R, Dekhuijzen PNR, Maltais F. L'atteinte du diaphragme et du quadriceps dans la BPCO : one manifestation systémique de la maladie ? Rev Fr Mal Resp 2011; 28: 1250.

FIGURES
REFERENCES
1. Celli BR, MacNee W. Standards for the diagnosis and treatment of patients with COPD a summary of the ATS/ERS position paper. Eur Respir J 2004; 23: 932.
2. Barnes PJ. Small airways in COPD. N Engl J Med 2004; 350: 26.
3. Hogg JC, Chu F, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med 2004; 350: 2645.
4. O'Donnell DE, Webb KA. Exertional breathlessness in patients with chronic airflow limitation. The role of lung hyperinflation. Am Rev Respir Dis 1993; 52: 70.
5. Barnes PJ, Celli BR. Systemic manifestations and comorbidities of COPD. Eur Respir J 2009; 33: 1165.
6. Agusti A, Calverley PM, Celli B et al. Characterisation of COPD heterogeneity in the ELIPSE cohort. Respir Res 2010; 11: 122.
7. McGarvey LP, Anderson JA et al. Ascertainment of cause-specific mortality in COPD: operations of the TORCH clinical endpoint committee. Thorax 2007; 62: 411.
8. Couillard A, Préfaut C. From muscle disuse to myopathy in COPD: potential contribution of oxydative stress. Eur Respir J 2005; 26: 703.
9. Serres I, Gautier V., Varray A., Préfaut C. Impaired squeletal muscle endurance related to physical inactivity and altered lung function in COPD patients. Chest 1998; 113: 900.
10. Marquis K, Jobin J, Maltais F et al. Mid thigh muscle cross-sectional area is a better predictor of mortality than body mass index in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2002; 166: 809.
11. Pitta F., Troosters T., Decramer M. et al. Characteristics of physical activities in daily life in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2005; 171: 972.
12. Couillard A, Maltais F, Préfaut C et al. Exerciseinduced quadriceps oxydative stress and peripheral muscle dysfonction in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2003; 167: 1664.
13. Koechlin C, Couillard A, Préfaut C et al. Does oxydative stress alter quadriceps endurance in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2004; 169: 1022.
14. Caron MA, Debigaré R, Dekhuijzen PNR, Maltais F. L'atteinte du diaphragme et du quadriceps dans la BPCO : one manifestation systémique de la maladie ? Rev Fr Mal Resp 2011; 28: 1250.
ARTICLE INFO
DOI: 10.12699/jfvp.4.13.2013.6
Conflict of Interest
Non
Date of manuscript receiving
26/04/2013
Date of publication after correction
15/10/2013
Article citation
Préfaut C. Physiopathology of chronic obstructive pulmonary disease - COPD. J Func Vent Pulm 2013;04(13):6-13.



Copyright: jfvpulm.com