
 English
English
 French
French
 Pubmed
Pubmed Google Scholar
Google Scholar Cross Ref
Cross Ref Visistor: 1021
Visistor: 1021
Les manifestations thoraciques de la drépanocytose
Thoracic manifestations of sickle cell disease
R. Bouchentouf
Service de Pneumologie, Hôpital Militaire Avicenne, Marrakech
Laboratoire PCIM, FMPM, Université Cadi Ayyad , Marrakech. Maroc
Corresponding author
Pr. Rachid BOUCHENTOUF
Service de Pneumologie
Hôpital Militaire Avicenne Marrakech
12 Bb la Resistance, Marrakech 40000. Maroc
E-mail: bouchentouf_rachid@yahoo.fr
ABSTRACT
Sickle cell disease is the most frequent hemoglobinopathy, is an autosomal genetic disease with transmission recessive, frequently observed in black patients.
The thoracic manifestations are various dominated by acute chest syndrome and the osseous vaso-occlusive crisis. These complications remain in the front causes of the death in sickle cell disease’s patients. Other chronic respiratory manifestations and cardiac and bone complication can be observed.
A better understanding of pathophysiological mechanisms responsible for those complications is necessary to develop new preventive and therapeutic approaches.
KEYWORDS: Sickle cell disease, thorax, manifestations
RÉSUMÉ
La drépanocytose est l’hémoglobinopathie la plus fréquente, c’est une maladie génétique à transmission autosomique récessive fréquente chez les sujets de race noire.
Les manifestations thoraciques sont polymorphes dominées par le syndrome thoracique aigu et les crises osseuses vaso-occlusives. Ces complications restent au premier rang des causes de mortalité des patients drépanocytaires. D’autres complications pulmonaires chroniques, cardiaques et ostéo-articulaires peuvent être rencontrées.
Une meilleure compréhension des mécanismes physiopathologiques responsables de ses manifestations naîtront de nouvelles approches préventives et thérapeutiques efficaces.
MOTS CLES: Drépanocytose, thorax, manifestations
INTRODUCTION
La drépanocytose est l’hémoglobinopathie la plus fréquente au monde survenant essentiellement chez les sujets de race noire. Il s’agit d’une maladie génétique caractérisée par la présence d’une hémoglobine S qui entraîne dans certaines conditions une perte de la plasticité des hématies responsable en partie des manifestations cliniques de la maladie [1].
Environ 15 % des patients atteints de ce syndrome développent une maladie grave. Néanmoins il faut souligner que tous les patients, y compris ceux chez qui l’évolution de la maladie paraît bénigne, sont exposés à la survenue brutale et imprévisible de complications pouvant engager le pronostic vital. Les causes de mortalité sont les crises vaso-occlusives osseuses et les syndromes thoraciques aigus.
COMPLICATIONS PULMONAIRES
Les complications pulmonaires sont les déterminants majeurs de la morbidité et de la mortalité de la maladie, elles peuvent être aigues ou chroniques [2].
Complications aigues: Syndrome Thoracique Aigu (STA; Acute Chest Syndrome)
Définition. Le STA est défini par l’association de fièvre ou de douleur thoracique à des anomalies radiologiques récentes, de type infiltrats alvéolaires englobant au moins un segment. STA est plus fréquent chez les patients homozygotes, mais reste possible dans les formes hétérozygotes, et peu être inaugurale de la maladie drépanocytaire. Certaines situations comme la grossesse (en particulier pendant la période du post-partum) ou les interventions chirurgicales en majorent le risque de survenue [3]. Le STA est la principale cause de décès et la deuxième cause d’hospitalisation des patients drépanocytaires [4].
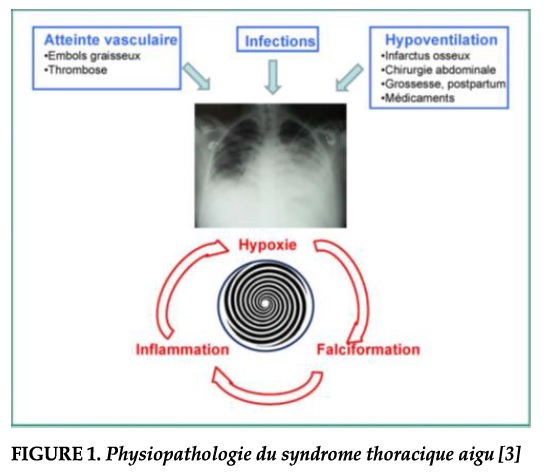
Physiopathologie. La physiopathologie du STA fait intervenir des phénomènes de vaso-occlusion au niveau de la circulation pulmonaire. Ces phénomènes sont déclenchés par diverses circonstances cliniques favorisantes, ses principales étiologies sont l’infection, l’embolie graisseuse, et l’hypoventilation alvéolaire. Les études les plus récentes utilisant les moyens de diagnostic modernes, et en particulier la fibroscopie bronchique avec prélèvements bactériologiques protégés, ont montre que l ‘infection n’explique que 10 a 20% des épisodes de syndrome thoracique [6]. Il a été démontré précédemment, grâce à I’utilisation du lavage broncho-alvéolaire (LBA), avec une technique de coloration particulière permettant la mise en évidence des graisses (coloration oil red 0) que près de la moitie des syndromes thoraciques seraient dus à des embolies pulmonaires graisseuses provenant d’infarctus ostéo-médullaires [7]. Les mécanismes physiopathologiques impliquées dans ces événements sont de mieux en mieux comprises et incluent entres autres l’obstruction mécanique par des hématies rigides et déformées (falciformées) mais aussi des phénomènes plus complexes d’adhérence des globules rouges et des leucocytes aux parois vasculaires, des phénomènes inflammatoires, et l’hypoxémie formant ainsi un cercle vicieux (Figure 1) [3].
Diagnostic. Le syndrome thoracique aigu se manifeste cliniquement par une douleur thoracique, de la fièvre, des râles crépitants ou une diminution du murmure vésiculaire [8]. À la radiologie, on note des images alvéolaires, multilobaires, souvent associées à une atteinte pleurale, l’atteinte est le plus souvent bilatérale, prédominant aux deux lobes inférieurs. Une radiographie initiale normale ne permet pas d’écarter un syndrome thoracique du fait du retard radiologique. La tomodensitométrie thoracique, réalisée dans les formes graves, évalue l’étendue de l’atteinte parenchymateuse [9]. La biologie est inutile au diagnostic, mais le dosage des gaz du sang est nécessaire pour évaluer la gravité; il existe souvent une hyperleucocytose et une augmentation de la CRP quasi constante et non synonyme d’infection bactérienne.
Traitement. Le traitement du STA est surtout symptomatique; il vise à lutter contre les phénomènes vaso-occlusifs en se basant sur l’hydratation, l’oxygénothérapie et un choix judicieux des antalgiques pour traiter les douleurs, tout en préservant la ventilation alvéolaire [2]. On y adjoint systématiquement une antibiothérapie probabiliste couvrant le pneumocoque et les germes encapsulés (amoxicilline ± macrolides ou fluoroquinolones) [10]. Tout critère de gravité doit conduire à intensifier la surveillance clinique et faire discuter un transfert en soins intensifs, et la réalisation en l’absence de contre-indication d’un échange transfusionnel qui doit parfois être répété. Un traitement étiologique doit être envisagé quand la cause est identifiée. La survenue d’un syndrome thoracique aigu doit faire discuter la mise en route d’un traitement de fond (hydroxycarbamide ou plus rarement un programme transfusionnel) dans le but de prévenir la récidive du syndrome thoracique aigu et de la crise vaso-occlusive, et de diminuer la mortalité [10].
Pronostic
Ce syndrome est la première cause de mortalité, il est responsable de 15 à 25% des décès chez ces patients [11,12]. Lors d’un syndrome thoracique aigu, la recherche de signes de gravité doit être immédiate et surtout répétée, étant donné le potentiel évolutif très rapide (quelques heures) vers un syndrome de détresse respiratoire aigu (Tableau 1) [4].
Complications pulmonaires chroniques
Poumon chronique du patient drépanocytaire. Cette complication est décrite depuis de nombreuses années, et serait secondaire à la constitution progressive d’une vasculopathie par falciformation intravasculaire pulmonaire. La symptomatologie clinique n’est pas spécifique, avec des douleurs thoraciques, une dyspnée de repos ou d’effort. Les épreuves fonctionnelles respiratoires (EFR) sont le marqueur le plus précoce et le plus fiable de l’atteinte chronique du poumon avant les premiers symptômes cliniques. L’évolution peut se faire vers l’insuffisance respiratoire chronique associant hypoxémie, fibrose interstitielle diffuse, cœur pulmonaire et syndrome restrictif.
Anomalies de la fonction pulmonaire. Un certain nombre d’anomalies ont été décrites chez les patients porteurs d’une drépanocytose sans pathologie interstitielle décelable sur l’imagerie. Chez un grand nombre de patients il semble exister soit une obstruction bronchique ou un syndrome restrictif modéré avec baisse modérée de la DLCO [13,14].
Autres complications chroniques
Les autres complications chroniques à rechercher sont l’asthme présent chez 5% des adultes, l’hypoxémie nocturne qui peut être un facteur favorisant d’AVC ou de crises vaso-occlusives [15]. En dehors de l’hypoxémie isolée, certains patients peuvent développer un vrai syndrome d’apnée du sommeil obstructif [3].
Traitement. Le traitement du STA est surtout symptomatique; il vise à lutter contre les phénomènes vaso-occlusifs en se basant sur l’hydratation, l’oxygénothérapie et un choix judicieux des antalgiques pour traiter les douleurs, tout en préservant la ventilation alvéolaire [2]. On y adjoint systématiquement une antibiothérapie probabiliste couvrant le pneumocoque et les germes encapsulés (amoxicilline ± macrolides ou fluoroquinolones) [10]. Tout critère de gravité doit conduire à intensifier la surveillance clinique et faire discuter un transfert en soins intensifs, et la réalisation en l’absence de contre-indication d’un échange transfusionnel qui doit parfois être répété. Un traitement étiologique doit être envisagé quand la cause est identifiée. La survenue d’un syndrome thoracique aigu doit faire discuter la mise en route d’un traitement de fond (hydroxycarbamide ou plus rarement un programme transfusionnel) dans le but de prévenir la récidive du syndrome thoracique aigu et de la crise vaso-occlusive, et de diminuer la mortalité [10].
Pronostic. Ce syndrome est la première cause de mortalité, il est responsable de 15 à 25% des décès chez ces patients [11,12]. Lors d’un syndrome thoracique aigu, la recherche de signes de gravité doit être immédiate et surtout répétée, étant donné le potentiel évolutif très rapide (quelques heures) vers un syndrome de détresse respiratoire aigu (Tableau 1) [4].
LES ATTEINTES CARDIAQUES
Peu d’études sur l’atteinte cardiaque sont disponibles, néanmoins schématiquement on distingue trois types d’atteintes surtout chez les sujets homozygotes.
Hypertension artérielle pulmonaire
L’HTP est une complication des anémies hémolytiques héréditaires décrite depuis de nombreuses années. L’hypertension artérielle pulmonaire (HTAP) pré capillaire est une complication relativement peu fréquente de cette maladie (de l’ordre de 3 à 4% des patients) [16]. Elle se caractérise par un profil hémodynamique différent de l’HTAP idiopathique avec des niveaux de pressions pulmonaires et de résistances vasculaires pulmonaires beaucoup plus basses [16]. Néanmoins, ce type d’atteinte vasculaire pulmonaire semble avoir un impact important sur le statut fonctionnel et le pronostic vital des patients drépanocytaires. Les traitements ne sont pas encore bien codifiés, et une attention particulière est nécessaire en cas d’emploi du sildénafil, qui peut provoquer des crises vaso-occlusives [17]. La prévalence élevée de l’HTAP d'une part et son impact global sur la mortalité justifient un dépistage systématique une échocardiographie par voie trans-thoracique, à distance d'une crise vaso-occlusive, à la recherche d'une fuite tricuspide avec calcul de la vitesse de régurgitation en doppler [18].
Anomalies des cavités gauches liées à l'anémie chronique
Les patients drépanocytaires du fait de leur anémie chronique ont un débit et un index cardiaque au repos augmenté de 30 à 50% comparativement à des sujets sains du même âge [18]. Des souffles systoliques éjectionnels ou d’insuffisance mitrale fonctionnelle, une hyperpulsatilité artérielle, des signes d’hypertrophie ventriculaire gauche peuvent être constatés chez le drépanocytaire.
Cardiomyopathie liée à la surcharge en fer
L’hémochromatose une source non négligeable de morbimortalité chez les patients drépanocytaires justifiant de transfusions répétées [19]. La surcharge de fer peut causer une cardiomyopathie avec au début les signes cliniques d'insuffisance cardiaque congestive qui sont non spécifiques, tout comme les éventuelles anomalies à I' ECG, à un stade tardif elle réalise un aspect de cardiomyopathie restrictive avec dysfonction systolique du VG de sévérité variable selon l'ancienneté et l'intensité de la surcharge en fer. L'étude en mode écho-doppler tissulaire permet de détecter des signes d’atteintes plus précoces (trouble de la motilité septale) [20], mais l’IRM cardiaque en mode T2 est loin l’examen le plus sensible pour le dépistage et la quantification de la surcharge en fer du myocarde [21].
Les atteintes Osseuses
Les atteintes ostéoarticulaires occupent une place importante dans les manifestations cliniques de la drépanocytose, elles peuvent avoir une expression polymorphe dominée par les crises vaso-oclusives et les infections ostéo-articulaires. Une crise vaso-occlusive ne doit jamais être considérée comme banale car elle peut rapidement se compliquer d’un syndrome thoracique aigu, d’une défaillance multi viscérale ou d’un sepsis, et conduire au décès [4]. La crise vaso-occlusive osseuse se manifeste par des douleurs osseuses aiguës très intenses (comparables à une fracture osseuse) touchant les métaphyses et les diaphyses des os longs, puis par ordre décroissant les vertèbres, les côtes, et les os du crâne [4]. L’évolution d’une crise vaso-occlusive osseuse est habituellement favorable en quelques jours. Les infections ostéo-articulaires sont pratiquement toujours des ostéomyélites alors que les arthrites septiques vraies sont rares. Le tableau clinique est sensiblement le même que celui des crises vaso-occlusives; la distinction est indispensable car le diagnostic d’ostéomyélite nécessite une antibiothérapie en urgence. La prolongation de la crise douloureuse au delà de deux semaines doit faire évoquer le diagnostic d’ostéomyélite, la biologie n’est d’aucun secours pour pouvoir faire la différence avec les crises vaso-occlusives [22]. Les radiographies des os douloureux ne permettent pas toujours de distinguer à un stade précoce une crise vaso-occlusive d’une ostéomyélite [23].
CONCLUSION
La drépanocytose est une maladie à laquelle le pneumologue peut être confronté en raison des complications pulmonaires aiguës ou chroniques. On doit craindre tout patient drépanocytaire consultant aux urgences la présence d’un syndrome thoracique aigu ou d’une infection, qu’il faut évaluer d’emblée la gravité et discuter l’indication d’échange transfusionnel.
CONFLIT D’INTÉRÊTS
Les auteurs ne déclarent pas de conflit d’intérêt.
REFERENCES
1. Aquino SL, Gamsu G, Fahy JV, et al. Chronic pulmonary disorders in sickle cell disease: findings at thin-section CT. Radiology 1994;193: 807-11.
2. Fauroux B, Muller MH, et al. Le poumon drépanocytaire de l’enfant à l’adulte. Rev Mal Resp 1998;15 :159-68.
3. Maître B, Mekontso-Dessap A, Habibi A, et al. Complications pulmonaires des syndromes drépanocytaires majeurs chez l’adulte. Rev Mal Resp 2011;28, 129 -137.
4. Gellen-Dautremer J, Brousse V, Arlet JB. Complications aiguës de la drépanocytose. Rev Prat 2014;64 :1114-19.
5. El Mekki F, Taktak S, Khaldi H, et al. Syndrome thoracique aigu révélateur d’une drépanocytose: À propos d’un cas avec revue de la littérature. Rev Pneumol Clin 2006;62:195-9.
6. Bachir D. Drépanocytose. Rev Fran Lab 2000;324 :29-35.
7. Godeau B, Schaeffer A, Bachir D , et al. Broncho alveolar lavage in adult sickle cell patients with acute chest syndrome: value for diagnostic assessment of fat embolism, Am J Resp Crit Care Med.1996;153:1691- 96.
8. Vichinsky EP, Styles LA, Colangelo LH, et al. Acute chest syndrome in sickle cell disease: clinical presentation and course. Blood 1997;89:1787-92..
9. Mekontso Dessap A, Deux JF, Habibi A, et al. Lung imaging during acute chest syndrome in sickle cell disease: computed tomography patterns and diagnostic accuracy of bedside chest radiograph. Thorax 2014; 69:144-51.
10. Lionnet F, Arlet JB, Bartolucci P, et al. Guidelines for management of adult sickle cell disease. Rev Med Int 2009; 30: S162-223.
11. Perronne V, Roberts-Harewood M, Bachir D, et al. Patterns of mortality in sickle cell disease in adults in France and England. Hematol J 2002;3:56-60.
12. Hamideh D, Alvarez O. Sickle cell disease related mortality in the United States (1999-2009). PediatrBlood Cancer2013;60: 1482 -6.
13. Klings ES, Wyszynski DF, Nolan VG, Steinberg MH. Abnormal pulmonary function in adults with sickle cell anemia. Am J Respir Crit Care Med 2006; 173: 1264-9.
14. MacLean JE , Atenafu E , Kirby-Allen M, et al. Longitudinal decline in lung volume in a population of children with sickle cell disease. Am J Respir Crit Care Med 2008;178:1055-9.
15. Zaïen I, Maître B. Drépanocytoses et complications pulmonaires. Rev Mal Resp Actualités (2015) 7, 89-91.
16. Savale L , Maitre B, Bachir D, et al. Hypertension artérielle pulmonaire et drépanocytose. Presse Med. 2013; 42: 338-346.
17. Machado RF, Barst RJ, Yovetich NA, et al. Hospitalization for pain in patients with sickle cell disease treated with sildenafil for elevated TRV and low exercise capacity. Blood 2011; 118: 855-64.
18. Michel M. Atteintes cardiaques au cours de la drépanocytose. Arch Malad Coeur Vais Prat 2007:18-19.
19. Darbari DS, et al. Circumstances of death in adults with sickle cell anemia. Am J Hematol 2006; 81: 858- 63.
20. Vogel M, et al. Tissue Doppler echocardiography in patients with thalassaemia detects early myocardial dysfunction related to rnyocardial iron overload. Eur Heart J 2003; 24:113-19.
21. Anderson U et al. Cardiovascular T2-star (T2*) rnagnetic resonance for the early diagnosis of myocardial iron overload. Eur Heart J 2001; 22: 2171-79.
22. Mary P. Complications ostéo-articulaires de la drépanocytose. Archives de Pédiatrie 2008 ;15: 639-641.
23. Jean Baptiste G, De Ceulaer K. Actualité des manifestations rhumatologiques des hémoglobinopathies. Rev Rhum2003; 157-61

FIGURES/TABLES

REFERENCES
1. Aquino SL, Gamsu G, Fahy JV, et al. Chronic pulmonary disorders in sickle cell disease: findings at thin-section CT. Radiology 1994;193: 807-11.
2. Fauroux B, Muller MH, et al. Le poumon drépanocytaire de l’enfant à l’adulte. Rev Mal Resp 1998;15 :159-68.
3. Maître B, Mekontso-Dessap A, Habibi A, et al. Complications pulmonaires des syndromes drépanocytaires majeurs chez l’adulte. Rev Mal Resp 2011;28, 129 -137.
4. Gellen-Dautremer J, Brousse V, Arlet JB. Complications aiguës de la drépanocytose. Rev Prat 2014;64 :1114-19.
5. El Mekki F, Taktak S, Khaldi H, et al. Syndrome thoracique aigu révélateur d’une drépanocytose: À propos d’un cas avec revue de la littérature. Rev Pneumol Clin 2006;62:195-9.
6. Bachir D. Drépanocytose. Rev Fran Lab 2000;324 :29-35.
7. Godeau B, Schaeffer A, Bachir D , et al. Broncho alveolar lavage in adult sickle cell patients with acute chest syndrome: value for diagnostic assessment of fat embolism, Am J Resp Crit Care Med.1996;153:1691- 96.
8. Vichinsky EP, Styles LA, Colangelo LH, et al. Acute chest syndrome in sickle cell disease: clinical presentation and course. Blood 1997;89:1787-92..
9. Mekontso Dessap A, Deux JF, Habibi A, et al. Lung imaging during acute chest syndrome in sickle cell disease: computed tomography patterns and diagnostic accuracy of bedside chest radiograph. Thorax 2014; 69:144-51.
10. Lionnet F, Arlet JB, Bartolucci P, et al. Guidelines for management of adult sickle cell disease. Rev Med Int 2009; 30: S162-223.
11. Perronne V, Roberts-Harewood M, Bachir D, et al. Patterns of mortality in sickle cell disease in adults in France and England. Hematol J 2002;3:56-60.
12. Hamideh D, Alvarez O. Sickle cell disease related mortality in the United States (1999-2009). PediatrBlood Cancer2013;60: 1482 -6.
13. Klings ES, Wyszynski DF, Nolan VG, Steinberg MH. Abnormal pulmonary function in adults with sickle cell anemia. Am J Respir Crit Care Med 2006; 173: 1264-9.
14. MacLean JE , Atenafu E , Kirby-Allen M, et al. Longitudinal decline in lung volume in a population of children with sickle cell disease. Am J Respir Crit Care Med 2008;178:1055-9.
15. Zaïen I, Maître B. Drépanocytoses et complications pulmonaires. Rev Mal Resp Actualités (2015) 7, 89-91.
16. Savale L , Maitre B, Bachir D, et al. Hypertension artérielle pulmonaire et drépanocytose. Presse Med. 2013; 42: 338-346.
17. Machado RF, Barst RJ, Yovetich NA, et al. Hospitalization for pain in patients with sickle cell disease treated with sildenafil for elevated TRV and low exercise capacity. Blood 2011; 118: 855-64.
18. Michel M. Atteintes cardiaques au cours de la drépanocytose. Arch Malad Coeur Vais Prat 2007:18-19.
19. Darbari DS, et al. Circumstances of death in adults with sickle cell anemia. Am J Hematol 2006; 81: 858- 63.
20. Vogel M, et al. Tissue Doppler echocardiography in patients with thalassaemia detects early myocardial dysfunction related to rnyocardial iron overload. Eur Heart J 2003; 24:113-19.
21. Anderson U et al. Cardiovascular T2-star (T2*) rnagnetic resonance for the early diagnosis of myocardial iron overload. Eur Heart J 2001; 22: 2171-79.
22. Mary P. Complications ostéo-articulaires de la drépanocytose. Archives de Pédiatrie 2008 ;15: 639-641.
23. Jean Baptiste G, De Ceulaer K. Actualité des manifestations rhumatologiques des hémoglobinopathies. Rev Rhum2003; 157-61
ARTICLE INFO
DOI: 10.12699/jfvp.6.19.2015.13
Conflict of Interest
Non
Date of manuscript receiving
22/3/2015
Date of publication after correction
14/10/2015
Article citation
Bouchentouf R. Thoracic manifestations of sickle cell disease. J Func Vent Pulm 2015;19(6):13-16.



Copyright: jfvpulm.com