
 English
English
 French
French
 Pubmed
Pubmed Google Scholar
Google Scholar Cross Ref
Cross Ref Visistor: 1590749
Visistor: 1590749The antisynthetase syndrome: ‘‘welcome your patients and you will have the diagnosis’’
Le syndrome des antisynthétases: ‘‘saluez vos patients et vous en aurez le diagnostic’’
K. Chaker, S. Khairallah, O. Iziki, H. Tahouna, M. Herrag
Service de Pneumologie. Département des Maladies Respiratoires
Centre Hospitalier Universitaire Mohammed VI. Faculté de Médecine et de Pharmacie Oujda - Maroc
Corresponding author
Dr. Khalid CHAKER
Service de Pneumologie
Département des Maladies Respiratoires Centre Hospitalier Universitaire Mohammed VI
Faculté de Médecine et de Pharmacie Oujda - Maroc
Adresse e-mail: dr_khalid@live.fr
ABSTRACT
Introduction. The anti-synthetase syndrome is defined by the association between inflammatory myopathy, interstitial lung disease, polyarthritis, Raynaud syndrome, and skin abnormalities like “mechanic hands”, which is associated with the presence of specific auto-antibodies, anti-aminoacyl-ARNt-syntheases. Among it, the anti-Jo1 antibody is the most frequent.
The interstitial lung damage is important because it conditions the vital prognostic.
Observation. A 60 years man who suffered since 20 years a dyspnea at exercise, associated with polyarthritis, myalgia, and Raynaud phenomenon. The clinical examination in this patient founded out a hyperkeratosis fissure in his hands. The diagnosis of antisynthetase syndrome was evoked and confirmed by the presence of anti-JO1 antibodies. The patient was treated by high dose of corticosteroids and needed then the association with immunosupressor in following year with satisfactory evolution.
Conclusion. The clinical examination is fundamental for the diagnosis of this syndrome. This syndrome is still unclear with late diagnosis by finding the aspect of mechanic hands.
KEYWORDS: Inflammatory myopathy, antisynthetase, diffuse infiltrative lung disease
RÉSUMÉ
Introduction. Le syndrome des antisynthétases est définit par l’association d’une myopathie inflammatoire, à une pneumopathie interstitielle, une polyarthrite, un phénomène de Raynaud et des anomalies cutanées à type de «mains de mécaniciens», avec la présence d’auto-anticorps spécifiques, les anti-aminoacyl-ARNt-synthétases dont le plus fréquent est l’anticorps anti-Jo1.
L’atteinte pulmonaire interstitielle est importante car elle conditionne le pronostic vital.
Observation. C’est une patiente de 60 ans qui souffrait depuis vingt ans d’une dyspnée d’effort associée à des polyarthralgies, des myalgies et un phénomène de Raynaud, chez qui l’examen clinique objectivait une hyperkératose fissuraire des mains. Le diagnostic d’un syndrome des antisynthétases était évoqué et confirmé par la présence des anticorps anti-JO1. La patiente était mise sous corticothérapie à forte dose, nécessitant une année après l’association d’un immunosuppresseur avec une évolution satisfaisante.
Conclusion. L’examen clinique est fondamental pour évoquer ce syndrome, qui reste méconnu et de diagnostic tardif, en reconnaissant l’aspect des mains de mécanicien.
MOTS CLÉS: Myopathie inflammatoire, antisynthétase, pneumopathie infiltrative diffuse
INTRODUCTION
Le syndrome des antisynthétases (SAS) est une connectivite auto-immune hétérogène, décrit pour la première fois en 1989, comme l’association d’une myopathie inflammatoire avec une pneumopathie interstitielle diffuse (PID), une polyarthrite, un phénomène de Raynaud et une atteinte cutanée de type hyperkératose fissuraire des doigts appelée «mains de mécaniciens», avec la présence d’auto-anticorps spécifiques: les anti-aminoacyl-ARNt-synthétases (AAS) dont le plus fréquent est l’anticorps anti-Jo1 [1-4]. Il s’agit d’une entité rare dont les mécanismes immunologiques soient aujourd’hui mieux compris [2, 5].
A travers ce premier cas clinique diagnostiqué dans notre formation, nous insistons sur le rôle de l’examen clinique dans l’orientation diagnostique surtout en cas d’une PID inexpliquée.
OBSERVATION
C’est une patiente âgée de 60 ans, qui était admise pour une dyspnée d’effort associée à une altération modérée de l’état général. À l’anamnèse, la patiente se plaignait de polyarthralgies évoluant depuis 20 ans, associés à une fatigabilité musculaire et une difficulté à effectuer des gestes simples affectant la qualité de vie, un phénomène de Raynaud et une xérostomie sans xérophtalmie.
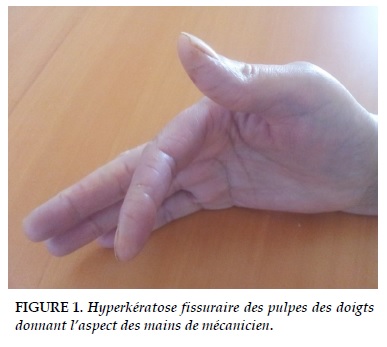
L’examen physique objectivait des râles crépitants bilatéraux, une hyperkératose fissuraire des mains donnant l’aspect des mains de mécanicien (Figure 1), une arthrite des poignets et chevilles, une sclérose cutanée de la face avec limitation de l’ouverture buccale.
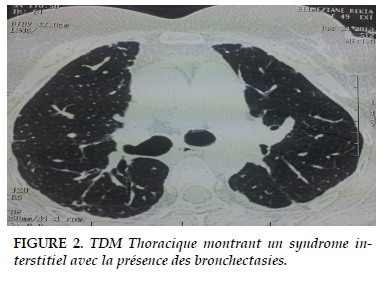
Le bilan biologique trouvait un syndrome inflammatoire, une élévation des enzymes musculaires, le bilan immunologique objectivait des AC anti-Jo1 positifs alors que les anticorps anti- nucléaires (AAN), facteurs rhumatoïdes et AC anti-Scl70 étaient négatifs. L’imagerie thoracique objectivait un syndrome interstitiel associé à des bronchectasies (Figure 2).
Les explorations fonctionnelles respiratoires (EFR) montraient un syndrome restrictif. La fibroscopie avec biopsie bronchique, la radiographie des mains et pieds ainsi que la biopsie des glandes salivaires étaient normaux. La patiente était mise sous-corticothérapie forte dose: 1 mg/kg par jour pendant 6 semaines puis dégression jusqu’à la dose d’entretien 10 mg/jour, l’évolution était marquée une année après par l’aggravation de la dyspnée et des polyarthralgies, ainsi la patiente était mise sous méthotrexate avec une amélioration clinique, radiologique et fonctionnelle.
DISCUSSION
Le SAS est une entité rare, sa prévalence est estimée à 1,5 cas pour 100 000 personnes. Son incidence varie de 1,2 à 2,5 nouveaux cas par million d’habitants [1, 2, 6]. Avec une prédominance féminine (sex-ratio: 3/1). Ce syndrome touche des patients âgés en moyenne de 50 ans, notre patiente était âgée de 60 ans. Le SAS est un syndrome très hétérogène, dont le diagnostic positif reste parfois difficile, ses manifestations cliniques sont variables (Tableau I). Les signes généraux sont inconstants et dominés par la fièvre [1, 7].
L’atteinte pulmonaire (75% des cas) est souvent au premier plan et conditionne le pronostic, il s’agit dans la majorité des cas d’une PID [2, 8, 9]. Deux modes d’installation sont possibles: aigu ou progressif avec des pronostics différents. Dans la forme aigue, les patients présentent une dyspnée sévère, associée à une fièvre, elle s’accompagne d’un «dommage alvéolaire diffus». Cette forme nécessite souvent une prise en charge en réanimation et elle est de mauvais pronostic. Dans la forme progressive, la dyspnée est principalement d’effort associée à une toux sèche et persistante, des râles crépitants et plus rarement un hippocratisme digital. Chez notre patiente, l’installation de l’atteinte respiratoire était progressive. Sur le plan fonctionnel, les EFR renseignent non seulement sur la sévérité de la PID mais également sur son pronostic, le paramètre le plus fiable est la capacité de diffusion du monoxyde de carbone (DLCO).
Trois principaux tableaux radiologiques sont décrits: la pneumopathie interstitielle non spécifique (PINS), la pneumopathie interstitielle commune et la pneumopathie organisée. Le LBA permet d’identifier le type d’atteinte histologique pulmonaire et surtout de préciser son stade évolutif. Une fois le diagnostic de PID est posé, l’objectif principal est d’en évaluer la sévérité et le pronostic à long terme. Ainsi, à côté du mode de survenue de la PID, l’existence d’une atteinte des muscles respiratoires et la survenue d’une pneumopathie d’inhalation sont également des facteurs de mauvais pronostic. Trois évolutions sous traitement sont rapportées: une résolution dans 20% des cas, une stabilité dans 60% des cas ou une aggravation dans 20% des cas [10-13].
L’atteinte musculaire (67% des cas) peut prendre plusieurs formes, allant de l’augmentation asymptomatique des enzymes musculaires, aux myalgies isolées voire à la myosite sévère avec déficit musculaire. Dans les formes peu sévères, le diagnostic peut être apporté par l’électromyogramme, l’imagerie par résonance magnétique (IRM) et la biopsie musculaire [1,2,9].
Les caractéristiques histologiques de la myosite du SAS répondent plus souvent aux critères de dermatomyosite (atrophie péri-fasciculaire) avec parfois un aspect particulier sous la forme d’une fragmentation et une infiltration inflammatoire du tissu conjonctif périmysial [14]. Généralement, la faiblesse musculaire s’améliore sous traitement, néanmoins, cette évolution est difficile à prédire et certains patients peuvent évoluer vers une forme chronique ou présenter des rechutes, elle est plutôt sévère dans les SAS avec anti-Jo-1 [15].
Chez notre patiente, l’atteinte musculaire dominait le tableau clinique, avec un retentissement important sur la qualité de vie, l’évolution était marquée par une nette amélioration après démarrage de traitement.
Les manifestations articulaires (57% des cas) sont hétérogènes et regroupent des polyarthralgies inflammatoires localisées aux petites articulations qui sont peu sévères, des polyarthrites non déformantes le plus souvent, mais parfois également érosives de pronostic fonctionnel plus réservé. De ce fait, une évaluation rigoureuse est importante et doit comporter la recherche de facteurs rhumatoïdes et d’anticorps anti-peptide-citrulliné (négatifs chez notre patiente).
En effet, il a été démontré au cours du SAS que ces anticorps pouvaient être positifs définissant ainsi une entité mixte, chevauchant avec la polyarthrite rhumatoïde [2,16]. Dans ce contexte précis, ou devant des symptômes rhumatologiques réfractaires au traitement, une échographie et/ou une IRM articulaire peuvent avoir un intérêt diagnostique et de suivi. Toutefois, la sévérité de ces formes articulaires est difficile à prédire et à prendre en charge spécifiquement.
L’atteinte cutanée est assez caractéristique et très évocatrice du SAS et représente un élément sémiologique d’orientation. Dans notre cas, c’était un élément clinique inhabituel et en même temps fondamental pour évoquer ce syndrome. Il s’agit d’une hyperkératose fissuraire des pulpes et des bords latéraux des doigts donnant l’aspect des « mains de mécanicien ». Cet aspect est fréquent au cours de SAS, sa prévalence est variable de 11 à 72% selon les séries, et il est important de savoir le reconnaitre, car il peut permettre le diagnostic surtout dans les formes amyopathiques. Il évolue de façon parallèle aux autres manifestations, et il n’a aucun traitement spécifique [1, 17].
Le phénomène de Raynaud peut être associé au SAS, avec une fréquence supérieure à 44 % des cas. Sur le plan physiopathologique, cette atteinte peut être expliquée par une activation des cellules endothéliales par les anti-Jo1. La capillaroscopie peut révéler une atteinte microvasculaire de type sclérodermique. La sévérité du phénomène de Raynaud varie selon les cas, et peut parfois engendrer des ulcères digitaux [1,2].
Sur le plan immunologique, ce sont les anticorps anti-cytoplasmiques dirigés contre les enzymes aminoacyl-t- RNA-synthétases (AAS) qui permettent d’évoquer et de confirmer le diagnostic. Les plus fréquents sont les anticorps anti-Jo1, les autres sont: PL7, PL12, OJ, EJ, KS, anti- JS, anti- ZO et anti- YRS. Plusieurs études ont démontré que le spectre clinique du SAS dépend de la spécificité de l’anticorps associé [6,9,12].
Ainsi, les patients avec anti-Jo1 présentaient un SAS plus diffus avec myosite, pneumopathie interstitielle, arthralgies, signes de sclérodermie systémique, tandis que les patients avec anti-PL12 ou anti-PL7 présentaient un SAS plus volontiers limité aux poumons.
Le traitement est basé sur les glucocorticoïdes à doses élevées pendant les premières 4 à 6 semaines pour atteindre le contrôle de la maladie, suivies d’une dégression progressive jusqu’à l’obtention de la dose minimale efficace pour maintenir la rémission. Parfois, le recours à un immunosuppresseur est nécessaire surtout en cas de développement d’une cortico-résistance ou d’une cortico-dépendance. Toutefois, il est délicat de savoir quel immunosuppresseur proposer, quand le débuter ou le modifier.
Actuellement, les immunosuppresseurs les plus souvent prescrits sont le cyclophosphamide, la ciclosporine, le mycophénolate mofétil, ou le tacrolimus [18, 19].
Chez notre patiente, l’évolution sous traitement immunosuppresseur associant corticoïdes et méthotrexate était marquée par une amélioration satisfaisante.
CONCLUSION
Le SAS est une forme rare et méconnue des myopathies, caractérisé le plus souvent par un retard diagnostic et une difficulté de prise en charge. La pneumopathie interstitielle est un des éléments de mauvais pronostic et elle constitue un déterminant majeur de morbidité et de mortalité. Le diagnostic peut être porté par l’examen clinique, en reconnaissant l’aspect des mains de mécanicien.
CONFLITS D’INTÉRÊTS
Aucun.
REFERENCES
1. Frikha F, Saidi N, Snoussi M, Ben Salah R, Ben Ayed M, Daoud E, Hentati Y, Makni S, Mnif Z, Boudawara T, Masmoudi H, Bahloul Z. Le syndrome des antisynthétases: à propos de quatre observations et revue de la littérature. Rev Pneumol Clin 2012 ; 68: 351-360.
2. Hervier B, Benveniste O. Phénotypes cliniques et pronostic du syndrome des antisynthétases. Rev Med Interne 2013, http://dx.doi.org/10.1016/j.revmed.2013.09.003.
3. Katzap E, Barilla-LaBarca ML, Marder G. Antisynthetase syndrome. Curr Rheumatol Rep 2011; 13: 175-181.
4. Shinjo SK, Levy-Neto M. Anti-Jo-1 antisynthetase syndrome. Rev Bras Reumatol 2010; 50: 492-500.
5. Lundberg IE, Helmers SB. The type I interferon system in idiopathic inflammatory myopathies. Autoimmunity 2010; 43: 239-43.
6. Brouwer R, Hengstman GJ, Vree Egberts W. Autoantibody profiles in the sera of European patients with myositis. Ann Rheum Dis 2001; 60: 116-123.
7. Hervier B, Devilliers H, Stanciu R, Meyer A, Uzunhan Y, Masseau A. Hierarchical cluster and survival analyses of antisynthetase syndrome: phenotype and outcome are correlated with anti-tRNA-synthetase antibody specificity. Autoimmun Rev 2012; 12: 210-7.
8. Labirua A, Lundberg IE. Interstitial lung disease and idiopathic inflammatory myopathies: progress and pitfalls. Curr Opin Rheumatol 2010; 22: 633-8.
9. Legout L, Fauchais A, Hachulla E. Le syndrome des anti-synthétases : un sous-groupe des myopathies inflammatoires à ne pas méconnaître. Rev Méd Interne 2002 ; 23 : 273-82.
10. Váncsa A, Csípo I, Németh J, Dévényi K, Gergely L, Dankó K. Characteristics of interstitial lung disease in SS-A positive/Jo-1 positive inflammatory myopathy patients. Rheumatol Int 2009; 29: 989-994.
11. Fathi M, Vikgren J, Boijsen M, Tylen U, Jorfeldt L, Tornling G. Interstitial lung disease in polymyositis and dermatomyositis: longitudinal evaluation by pulmonary function and radiology. Arthritis Rheum 2008; 59: 677-85.
12. Yousem SA, Gibson K, Kaminski N, Oddis CV, Ascherman DP. The pulmonary histopathologic manifestations of the anti-Jo-1 tRNA synthetase syndrome. Mod Pathol 2010; 23: 874-880.
13. Guglielmi S, Merz T.M, Gugger M, Suter C, Nicod L.P. Acute respiratory distress syndrome secondary to antisynthetase syndrome is reversible with tacrolimus. Eur Respir J 2008; 31: 213-217.
14. Mozaffar T, Pestronk A. Myopathy with anti-Jo-1 antibodies: pathology in perimysium and neighbouring muscle fibres. J Neurol Neurosurg Psychiatry 2000; 68: 472-8.
15. Aggarwal R, Oddis CV. Therapeutic advances in myositis. Curr Opin Rheumatol 2012; 24: 635-41.
16. Park CK, Kim TJ, Cho YN, Kim IS, Lee HJ, Lee KE. Development of anti-synthetase syndrome in a patient with rheumatoid arthritis. Rheumatol Int 2011; 31: 529-32.
17. Bachmeyer C, Tillie-Leblond I, Lacert A, Cadranel J, Aractingi S. Mechanic’shands: a misleading cutaneous sign of the antisynthetase syndrome. Br J Dermatol 2007; 156: 192-4.
18. Zappa MC, Trequattrini T, Mattioli F et al. Rituximab treatment in a case of antisynthetase syndrome with severe interstitial lung disease and acute respiratory failure. Multidisciplinary Respiratory Medicine 2011; 6: 183-188.
19. Takada K, Nagasaka K, Mijasaka N. Polymyositis/dermatomyositis and interstitial lung disease: a new therapeutic approach with T-cell-specific immunosuppressants. Autoimmunitiy 2005; 38: 383-392.

FIGURES
REFERENCES
1. Frikha F, Saidi N, Snoussi M, Ben Salah R, Ben Ayed M, Daoud E, Hentati Y, Makni S, Mnif Z, Boudawara T, Masmoudi H, Bahloul Z. Le syndrome des antisynthétases: à propos de quatre observations et revue de la littérature. Rev Pneumol Clin 2012 ; 68: 351-360.
2. Hervier B, Benveniste O. Phénotypes cliniques et pronostic du syndrome des antisynthétases. Rev Med Interne 2013, http://dx.doi.org/10.1016/j.revmed.2013.09.003.
3. Katzap E, Barilla-LaBarca ML, Marder G. Antisynthetase syndrome. Curr Rheumatol Rep 2011; 13: 175-181.
4. Shinjo SK, Levy-Neto M. Anti-Jo-1 antisynthetase syndrome. Rev Bras Reumatol 2010; 50: 492-500.
5. Lundberg IE, Helmers SB. The type I interferon system in idiopathic inflammatory myopathies. Autoimmunity 2010; 43: 239-43.
6. Brouwer R, Hengstman GJ, Vree Egberts W. Autoantibody profiles in the sera of European patients with myositis. Ann Rheum Dis 2001; 60: 116-123.
7. Hervier B, Devilliers H, Stanciu R, Meyer A, Uzunhan Y, Masseau A. Hierarchical cluster and survival analyses of antisynthetase syndrome: phenotype and outcome are correlated with anti-tRNA-synthetase antibody specificity. Autoimmun Rev 2012; 12: 210-7.
8. Labirua A, Lundberg IE. Interstitial lung disease and idiopathic inflammatory myopathies: progress and pitfalls. Curr Opin Rheumatol 2010; 22: 633-8.
9. Legout L, Fauchais A, Hachulla E. Le syndrome des anti-synthétases : un sous-groupe des myopathies inflammatoires à ne pas méconnaître. Rev Méd Interne 2002 ; 23 : 273-82.
10. Váncsa A, Csípo I, Németh J, Dévényi K, Gergely L, Dankó K. Characteristics of interstitial lung disease in SS-A positive/Jo-1 positive inflammatory myopathy patients. Rheumatol Int 2009; 29: 989-994.
11. Fathi M, Vikgren J, Boijsen M, Tylen U, Jorfeldt L, Tornling G. Interstitial lung disease in polymyositis and dermatomyositis: longitudinal evaluation by pulmonary function and radiology. Arthritis Rheum 2008; 59: 677-85.
12. Yousem SA, Gibson K, Kaminski N, Oddis CV, Ascherman DP. The pulmonary histopathologic manifestations of the anti-Jo-1 tRNA synthetase syndrome. Mod Pathol 2010; 23: 874-880.
13. Guglielmi S, Merz T.M, Gugger M, Suter C, Nicod L.P. Acute respiratory distress syndrome secondary to antisynthetase syndrome is reversible with tacrolimus. Eur Respir J 2008; 31: 213-217.
14. Mozaffar T, Pestronk A. Myopathy with anti-Jo-1 antibodies: pathology in perimysium and neighbouring muscle fibres. J Neurol Neurosurg Psychiatry 2000; 68: 472-8.
15. Aggarwal R, Oddis CV. Therapeutic advances in myositis. Curr Opin Rheumatol 2012; 24: 635-41.
16. Park CK, Kim TJ, Cho YN, Kim IS, Lee HJ, Lee KE. Development of anti-synthetase syndrome in a patient with rheumatoid arthritis. Rheumatol Int 2011; 31: 529-32.
17. Bachmeyer C, Tillie-Leblond I, Lacert A, Cadranel J, Aractingi S. Mechanic’shands: a misleading cutaneous sign of the antisynthetase syndrome. Br J Dermatol 2007; 156: 192-4.
18. Zappa MC, Trequattrini T, Mattioli F et al. Rituximab treatment in a case of antisynthetase syndrome with severe interstitial lung disease and acute respiratory failure. Multidisciplinary Respiratory Medicine 2011; 6: 183-188.
19. Takada K, Nagasaka K, Mijasaka N. Polymyositis/dermatomyositis and interstitial lung disease: a new therapeutic approach with T-cell-specific immunosuppressants. Autoimmunitiy 2005; 38: 383-392.
ARTICLE INFO
DOI: 10.12699/jfvp.6.17.2015.42
Conflict of Interest
Non
Date of manuscript receiving
12/08/2014
Date of publication after correction
23/02/2015
Article citation
Chaker K, Khairallah S, Iziki O, Tahouna H, Herrag M. The antisynthetase syndrome: ‘‘welcome your patients and you will have the diagnosis’’. J Func Vent Pulm 2015;17(6):42-46.



Copyright: jfvpulm.com