
 English
English
 French
French
 Pubmed
Pubmed Google Scholar
Google Scholar Cross Ref
Cross Ref Visistor: 1590745
Visistor: 1590745Molecular abnormalities and target therapy for non-small cell lung cancers Anormalies moléculaires et ciblage thérapeutique des cancers broncho-pulmonaires non à petites cellules
C. Ngo1, S. Duong-Quy1,2
1: Université Paris Descartes. Paris - France
2: Collège de Médecine de Lam Dong. Dalat - Viet Nam
Corresponding author
Pr. Sy DUONG-QUY
Collège de Médecine de Lam Dong
Hôpital Cochin. Université Paris Descartes
E-mail: sduongquy.jfvp@gmail.com
ABSTRACT
Lung cancer is the first leading cause of death due to cancer in France and in the world with nearly 30,000 deaths (20%) in 2012 in France. The 5-year survival is about 15% for all confounded stages, with 70% of patients are inoperable.
In men, the incidence of lung cancer is stable while it is continuing increase in female. The principal cause of lung cancer is due to smoking. Among these cancers, there are non-small cell lung carcinoma (NMCLC) (80%) and small cell lung carcinoma (MCLC) (20%). Among NSLC, different histological subtypes are described: adenocarcinoma, squamous cell carcinoma, large cell carcinoma and rarer entities (adenosquamous, sarcomatoid carcinoma ...).
The histological subtype of lung cancers determines the therapeutic choice. NSCLC, when operable, are treated by surgery whereas SCLC within chemotherapy. Among NSCLC, the type of chemotherapy also varies depending on whether a squamous cell carcinoma, adenocarcinoma or large cell carcinoma.
The emergence of targeted therapies aiming to specific molecular abnormalities of cancer cells has changed the role of the pathologist, adding to its diagnostic and prognostic missions, a therapeutic challenge to identify oncogenic mutations "driver" accessible to a targeted therapy.
KEYWORDS: NSCLC, MCLC, adenocarcinoma, squamous cell carcinoma, chemotherapy
RÉSUMÉ
Le cancer broncho-pulmonaire est la 1ère cause de décès par cancer en France et dans le monde, avec près de 30 000 décès (20%) en 2012 en France. La survie à 5 ans est de l’ordre de 15% tous stades confondus, avec 70% des patients qui sont non opérables.
Chez l’homme, l’incidence du cancer broncho-pulmonaire est stable tandis qu’elle ne cesse d’augmenter chez la femme. La principale cause de cancer broncho-pulmonaire est due au tabac. Parmi ces cancers, on distingue les carcinomes broncho-pulmonaires non à petites cellules (CBNPC) (80%) des carcinomes broncho-pulmonaires à petites cellules (CBPC) (20%). Parmi les CBNPC, on décrit différents sous-types histologiques: les adénocarcinomes, les carcinomes épidermoïdes, les carcinomes à grandes cellules et des entités plus rares (adénosquameux, carcinome sarcomatoïde...).
Le sous-type histologique des cancers broncho-pulmonaires conditionne le choix thérapeutique. Les CBNPC, lorsqu’ils sont opérables, sont traités par chirurgie alors que les CBPC relèvent de la chimiothérapie. Parmi les CBNPC, le type de chimiothérapie varie également selon qu’il s’agit d’un carcinome épidermoïde, d’un adénocarcinome ou d’un carcinome à grandes cellules.
L’émergence de thérapeutiques ciblées visant des anomalies moléculaires spécifiques des cellules cancéreuses a modifié le rôle du pathologiste, ajoutant à ses missions diagnostiques et pronostiques, un défi thérapeutique pour identifier des mutations oncogéniques « driver » accessibles à une thérapie ciblée.
MOTS CLÉS: CBNPC, CBPC, adénocarcinomes, carcinomes épidermoïdes, chimiothérapie
LES ENJEUX DE LA BIOLOGIE MOLECULAIRE EN ONCOLOGIE THORACIQUE
Le cancer broncho-pulmonaire est la 1ère cause de décès par cancer en France et dans le monde, avec près de 30 000 décès (20%) en 2012 en France. La survie à 5 ans est de l’ordre de 15% tous stades confondus, avec 70% des patients qui sont non opérables. Chez l’homme, l’incidence du cancer broncho-pulmonaire est stable tandis qu’elle ne cesse d’augmenter chez la femme.
La principale cause de cancer broncho-pulmonaire est due au tabac. Parmi ces cancers, on distingue les carcinomes broncho-pulmonaires non à petites cellules (CBNPC) (80%) des carcinomes broncho-pulmonaires à petites cellules (CBPC) (20%). Parmi les CBNPC, on décrit différents sous-types histologiques: les adénocarcinomes, les carcinomes épidermoïdes, les carcinomes à grandes cellules et des entités plus rares (adénosquameux, carcinome sarcomatoïde...).
Le sous-type histologique des cancers broncho-pulmonaires conditionne le choix thérapeutique. Les CBNPC, lorsqu’ils sont opérables, sont traités par chirurgie alors que les CBPC relèvent de la chimiothérapie. Parmi les CBNPC, le type de chimiothérapie varie également selon qu’il s’agit d’un carcinome épidermoïde, d’un adénocarcinome ou d’un carcinome à grandes cellules. L’émergence de thérapeutiques ciblées visant des anomalies moléculaires spécifiques des cellules cancéreuses a modifié le rôle du pathologiste, ajoutant à ses missions diagnostiques et pronostiques, un défi thérapeutique pour identifier des mutations oncogéniques « driver » accessibles à une thérapie ciblée. Ce concept d’addiction oncogénique suggère que ces mutations oncogéniques « driver » impliquant une anomalie d’une protéine, d’une voie de signalisation, du contrôle du cycle cellulaire ou de la régulation de l’apoptose, des processus induisant la mobilité, l’invasion ou la néo-angiogénèse suffisent à enclencher l’oncogénèse. La prescription de ces médicaments ciblant ces mutations est conditionnée par la présence de l’anomalie moléculaire cible qu’il faut rechercher en routine.
Pour soutenir le développement d’une médecine personnalisée et faciliter l’accès des thérapies ciblées aux patients qui peuvent en bénéficier, l’Institut national du cancer (INCa) a mis en place des plateformes de génétique moléculaire en France, lancé plusieurs programmes prospectifs portant sur des biomarqueurs émergents pouvant être la cible de nouvelles thérapies actuellement en cours de développement. La mise en place du programme pour un accès sécurisé à des thérapies ciblées innovantes (AcSé), lancé en 2013, vise à proposer à des patients en échec thérapeutique de nouvelles thérapies ciblant des altérations génétiques présentes dans leur tumeur, avec ou sans autorisation de mise sur le marché (AMM).
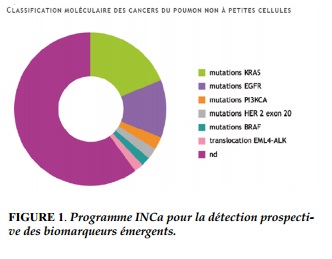
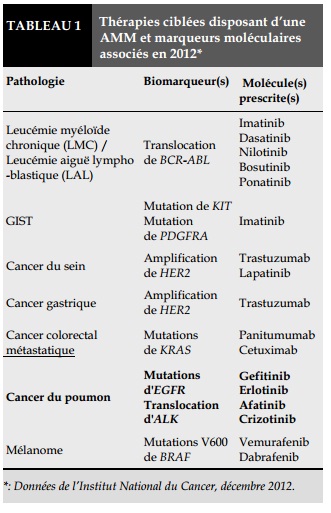
Actuellement, 60 tests moléculaires innovants sont disponibles. 8 biomarqueurs différents conditionnent l'emploi de molécules disposant d'une AMM dans 7 localisations tumorales, dont le cancer du poumon 3 (Tableau 1).
Ainsi, les altérations génétiques du gène du récepteur du facteur de croissance épidermique (EGFR) et d’ALK sont des biomarqueurs actuellement recherchés en routine, cibles de thérapeutiques ayant l’AMM. D’autres biomarqueurs émergents tels que KRAS, BRAF, PIK3CA, HER2, ROS1 et MET sont en cours d’évaluation du fait du développement rapide de thérapeutiques ciblées. Ces anomalies moléculaires sont mutuellement exclusives (Figure 1).
LES BIOMARQUEURS ACTUELS EN ONCOLOGIE THORACIQUE
Les mutations du récepteur de l’EGF
Structure, activation, voies de signalisation
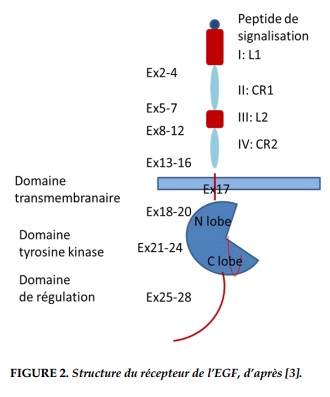
Le récepteur de l’EGF (EGFR) est un récepteur membranaire à activité tyrosine kinase, codé par un gène situé sur le chromosome 7p12. Protéine de 170kDa, elle appartient à la famille des récepteurs de l’EGF (ou ERBB/HER) dont font également partie les protéines ERBB2/HER2, ERBB3/HER3 et ERBB4/HER4. L’EGFR possède 4 domaines fonctionnels [1] (Figure 2): un domaine extracellulaire de liaison du ligand ; un domaine transmembranaire ; un domaine intracellulaire à activité tyrosine kinase et un domaine de régulation C-terminal.
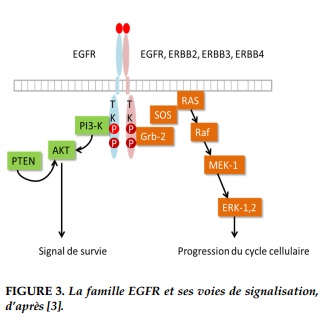
Il existe différents ligands connus: l’EGF (epidermal growth factor), le TGF-a (Transforming growth factor-alpha), l’amphiréguline et l’épiréguline. La fixation du ligand sur l’EGFR entraîne la formation d’un homo- ou hétéro-dimère, ce qui conduit à l’activation de l’EGFR par autophosphorylation du domaine tyrosine kinase. L’activation de l’EGFR induit différentes voies de signalisation dont la voie des phosphatidyl-inositol 3-kinases (PI3-K) et celle des MAP kinases qui contrôlent la prolifération cellulaire, la migration, l’échappement à l’apoptose, l’angiogénèse, processus impliqués dans la carcinogénèse1 (Figure 3).
La mutation de l’EGFR: une mutation clé
Les mutations oncogéniques activatrices de l’EGFR sont détectées avec une fréquence de 16,6% sur une cohorte de 2105 patients caucasiens (350 patients mutés EGFR) [2]. Ces mutations sont retrouvées sur les exons 18 à 21 qui codent pour le domaine tyrosine kinase de l’EGFR, au niveau du site de fixation de l’ATP qui est aussi le site de fixation des inhibiteurs de tyrosine kinase anti-EGFR (ITK-EGFR) ce qui confère une sensibilité aux ITK-EGFR [3]. Ces mutations de l’EGFR sont à l’origine de changements conformationnels et fonctionnels de l’EFGR entrainant une altération de la régulation on/off du récepteur en faveur d’une hyperactivation.
Elles sont plus fréquentes chez les femmes, d’origine asiatique et non fumeuses, en particulier dans les adénocarcinomes papillaires et lépidiques non mucineux (ex-bronchiolo-alvéolaires). Les mutations les plus fréquentes sont des délétions sur l’exon 19 (62%) ou des mutations ponctuelles faux-sens situées sur l’exon 21.
Les inhibiteurs de tyrosine kinase anti-EGFR
A ce jour, les deux ITK-EGFR ayant l’AMM européenne sont le gefitinib et l’erlotinib.
Le gefitinib (Iressa®) a obtenu une AMM en juin 2009 pour le traitement des patients avec une forme localement avancée ou métastatique de CBNPC et dont la tumeur porte une mutation activatrice de l'EGFR. Le gefitinib peut alors être utilisé en première ou en deuxième ligne de traitement.
L'erlotinib (Tarceva®) a obtenu une AMM en septembre 2011 pour le traitement en seconde ligne des patients avec une forme localement avancée ou métastatique de CBNPC:
Ainsi, l'essai de phase III IPASS a montré que le taux de réponse au gefitinib est de 71,2% chez les patients EGFR+ contre seulement 1,1% pour les patients EGFR- [4]. Pour les patients EGFR+, la survie sans progression est significativement plus longue pour les patients traités par gefitinib que pour ceux traités par chimiothérapie. Des résultats comparables ont été observés chez des patients traités par erlotinib [5].
Mécanismes de résistance aux inhibiteurs de tyrosine kinase anti-EGFR
Certaines mutations confèrent une résistance aux ITK-EGFR. Elles sont le plus souvent acquises au cours du traitement par ITK-EGFR. On connaît la mutation secondaire de résistance aux ITK située au niveau de l’exon 20 de l’EGFR, entraînant un remplacement d’une thréonine par une méthionine au niveau du codon 790 et conduisant à un changement de conformation du récepteur, empêchant la fixation des ITK-EGFR de 1ère génération [6]. Ce clone tumoral résistant apparaît sous ITK-EGFR chez environ 50% des patients porteurs d’un CBNPC EGFR+ [3]. Plusieurs ITK-EGFR de 2nde génération comme l’afatinib permettraient in vitro de passer outre cette résistance.
Techniques de détection des mutations EGFR
La technique de séquençage Sanger, qui étudie de façon exhaustive l’ADN à la recherche de mutations, est la technique de référence. Mais d’autres techniques d’analyse moléculaire ciblée telles que la PCR spécifique d’allèles sont des techniques plus sensibles et plus rapides. Cette analyse moléculaire peut se faire sur des biopsies, sur pièce opératoire ou prélèvement cytologique congelé ou inclus en paraffine. Elle nécessite des échantillons de bonne qualité (intérêt de la phase pré-analytique: cryopréservation rapide en azote liquide et fixation formolée de courte durée), avec un ratio cellule tumorale/tissu non tumoral d’au moins 15% (macrodissection sur lame si besoin pour éliminer des cellules stromales non tumorales).
Les réarrangements d’ALK
Réarrangements d’ALK et protéine de fusion
La protéine ALK est un récepteur à tyrosine kinase de la superfamille des récepteurs à l'insuline et dont le ligand est encore inconnu. Elle est codée par un gène situé sur le chromosome 2p23. Des altérations du gène ALK ont été mises en évidence dans différents cancers : lymphome anaplasique à grandes cellules, tumeur myofibroblastique et inflammatoire et le neuroblastome. En 2007, il a été démontré le rôle du réarrangement EML4 (Echinoderm microtubules associated protein like 4)-ALK (Anaplasic lymphoma kinase) dans le cancer broncho-pulmonaire, observé avec une fréquence de 6,7% dans les CBNPC [7]. La protéine de fusion EML4-ALK résulte d’une inversion du bras court du chromosome 2 aboutissant à la formation du gène de fusion EML4-ALK, mais d'autres translocations d’ALK sont possibles avec d’autres partenaires de fusion (TFG, KIF5B, KLC1).
Le plus souvent, les remaniements d'ALK aboutissent à une activation constitutive de la kinase ALK et des voies de signalisation en aval contrôlant la prolifération et la survie. Les altérations du gène ALK sont rares et retrouvées plus particulièrement chez les sujets jeunes, non ou peu tabagiques, à un stade avancé (métastases cérébrales). Elles sont décrites dans les adénocarcinomes TTF1+ le plus souvent, d’architecture tubulaire, papillaire, cribriforme ou solide en bague à chaton mais aussi dans les carcinomes épidermoïdes et adénosquameux.
Les inhibiteurs de tyrosine kinase anti-ALK
Le crizotinib est un inhibiteur de tyrosine kinase ciblant la protéine ALK mais aussi MET et ROS1. Lors d'un essai clinique de phase I, le crizotinib a démontré son efficacité pour le traitement en seconde ligne ou plus des patients dont la tumeur porte une translocation d'ALK. Après une durée moyenne de traitement de 6,4 mois, une réponse globale au crizotinib a été observée pour 57% des patients et une 33 % d'entre eux ont une stabilisation de la maladie [8]. Parmi la population de patients ALK+, la survie à 1 an est de 70% pour les patients sous crizotinib contre 44% pour le groupe contrôle et de 55% après 2 ans contre 12 % des patients du groupe contrôle [9].
Le Crizotinib (Xalkori®) a obtenu une AMM euro -péenne pour le traitement des patients atteints d'un CBNPC de type adénocarcinome avancé, chez des patients ALK+ prétraités, pour lesquels il n'existe pas d'alternative thérapeutique appropriée.
Techniques de détection des réarrangements ALK
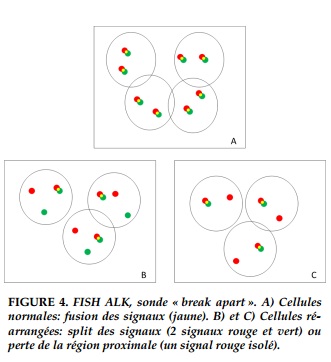
L’hybridation in situ en fluorescence (FISH) par sonde « break apart » est la technique de référence. Elle permet de visualiser les réarrangements de ALK indépendamment du partenaire de fusion, quelque soit le point de cassure chromosomique (Figure 4). Elle peut se faire sur tissu fixé en formol et inclus en paraffine. Cette technique coûteuse requiert une richesse tumorale sur la biopsie. L’interprétation est parfois délicate, le taux de cas non interprétable allant jusqu’à 19% [10].
L’immunohistochimie est une technique peu coûteuse avec une bonne sensibilité. Une immunohistochimie ALK+ suffit à conclure. En revanche, une immunohistochimie ALK- nécessite une confirmation par FISH.
La technique de RT-PCR, qui n’a pas encore de place en routine, nécessite une quantité de matériel tumoral importante. Sa sensibilité est assez bonne et permet surtout d’identifier les variants et les partenaires de fusion. Elle est utile en cas d’immunohistochimie et de FISH ALK non interprétables.
LES BIOMARQUEURS EMERGENTS EN ONCOLOGIE THORACIQUE
Mutations de KRAS
KRAS est une protéine à activité GTPase impliquée dans la signalisation des récepteurs membranaires, notamment l’EGFR. Les mutations de KRAS et EGFR sont mutuellement exclusives. Les mutations de KRAS sont associées à une résistance aux ITK-EGFR. La présence d’une mutation dans l’exon 2 de KRAS aboutit à la synthèse d’une protéine active constitutivement, une activation continue de la voie PI3-K, ce qui augmente la mobilité et la migration des cellules tumorales avec une diminution ou une perte d’expression de la E-cadhérine. Les mutations de KRAS sont observées avec une fréquence de 30% dans la population caucasienne, plus particulièrement chez des sujets fumeurs porteurs d’un adénocarcinome de type mucineux et plus rarement dans les carcinomes épidermoïdes. Des essais cliniques sont en cours avec des inhibiteurs de MAP kinases seuls ou en association aux inhibiteurs de mTOR ou à la chimiothérapie.
Amplification de MET
MET est un récepteur de tyrosine kinase, dont le gène est situé sur le même chromosome que l’EGFR (bras long du chromosome 7).
Le ligand est le facteur de croissance hépatocytaire (HGF, hepatocyte growth factor). L’activation de MET induit les mêmes voies de signalisation que l’EGFR. L’amplification de MET est rare (5% des CBNPC). L’augmentation du nombre de copies de MET dans les CBNPC, mise en évidence par FISH ou RT-PCR, est corrélée à un plus mauvais pronostic chez les patients atteints de CBNPC [11]. Elle est également impliquée dans les mécanismes de résistance acquise des cancers broncho-pulmonaires EGFR+ aux ITK-EGFR ; elle serait présente chez 5 à 20% des patients dans cette configuration [12].
Récemment a été ouvert en France le programme AcSé évaluant l’éfficacité du crizotinib dans les cancers broncho-pulmonaires amplifiés MET.
Translocation de ROS1
ROS1 est un récepteur à activité tyrosine kinase, codé par un gène situé sur le chromosome 6p21. La translocation de ROS1 a été décrite pour la première fois dans les glioblastomes. Elle est impliquée dans 1% des CBNPC, plus particulièrement chez les jeunes non fumeurs, dans les adénocarcinomes de même morphologie que ceux qui sont ALK+. Comme pour l’ampfication de MET, il s’agit d’une cible pour laquelle un abord thérapeutique existe dans le cadre du programme Acsé.
Mutations de BRAF
BRAF est une protéine kinase de la famille Raf qui régule les protéines MAPK et ERK et joue un rôle dans les processus de division et de différenciation cellulaire. Elle est notamment située en aval des voies de signalisation de l’EGFR et des protéines Ras. Les mutations de BRAF ont été mises évidence dans 40% des mélanomes et la mutation du codon 600 de BRAF est la plus fréquente, y compris dans le CBNPC (5% des adénocarcinomes bronchiques).
Des essais sont en cours pour tester l’efficacité des inhibiteurs de BRAF.
Amplifications et mutations d’HER2
HER2 est un autre membre de la famille des récepteurs de l’EGF. Dans le CBNPC, HER2 est surexprimée dans 17-42% des adénocarcinomes et 0-5% des carcinomes épidermoides. La surexpression d’HER2 est associée à un mauvais pronostic. Les mutations (insertions dans l’exon 20) d’HER2 sont rares, observées dans 1-2% des cas, plus fréquemment chez les femmes, non fumeuses, d’origine asiatique. L’analyse se fait par immunohistochimie et FISH (amplification) ou PCR (mutations exon 20).
Des études sont en cours pour tester l’efficacité des thérapies ciblant HER2 telles que le lapatinib, l’afatinib ou le trastuzumab.
Mutations et amplifications de PIK3CA
Les PI3 kinases sont des effecteurs clés des récepteurs de facteur de croissance. Le gène PIK3CA est muté ou amplifié dans de nombreux cancers.
Des thérapies ciblant les PI3 kinases sont en cours dans le cancer broncho-pulmonaire.
CONFLIT D’INTÉRÊTS
Aucun.
REFERENCES
1. Mitsudomi T, Yatabe Y. Epidermal growth factor receptor in relation to tumor development: EGFR gene and cancer. FEBS J 2010, 277: 301–308.
2. Rosell R, Moran T. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009;361(10):958-67.
3. Sharma SV, Bell DW. EGFR mutations in lung cancer. Nat Rev Cancer. 2007;7(3):169-81.
4. Mok TS, Wu YL. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009 Sep 3;361(10):947-57.
5. Rosell R, Carcereny E. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239-46.
6. Oxnard GR, Arcila ME. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res. 2011;17(6):1616-22.
7. Soda M, Choi YL. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561-6.
8. Kwak EL, Bang YJ. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363(18):1693-703.
9. Shaw AT, Yeap BY. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol. 2011;12(11):1004-12.
10. McLeer-Florin A, Moro-Sibilot D. Dual IHC and FISH testing for ALK gene rearrangement in lung adenocarcinomas in a routine practice: a French study. J Thorac Oncol. 2012;7(2):348-54.
11. Cappuzzo F, Marchetti A. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J Clin Oncol. 2009;27(10):1667-74.
12. Sequist LV, Waltman BA. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26.

FIGURES/TABLE
REFERENCES
1. Mitsudomi T, Yatabe Y. Epidermal growth factor receptor in relation to tumor development: EGFR gene and cancer. FEBS J 2010, 277: 301–308.
2. Rosell R, Moran T. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009;361(10):958-67.
3. Sharma SV, Bell DW. EGFR mutations in lung cancer. Nat Rev Cancer. 2007;7(3):169-81.
4. Mok TS, Wu YL. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009 Sep 3;361(10):947-57.
5. Rosell R, Carcereny E. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239-46.
6. Oxnard GR, Arcila ME. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res. 2011;17(6):1616-22.
7. Soda M, Choi YL. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561-6.
8. Kwak EL, Bang YJ. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363(18):1693-703.
9. Shaw AT, Yeap BY. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol. 2011;12(11):1004-12.
10. McLeer-Florin A, Moro-Sibilot D. Dual IHC and FISH testing for ALK gene rearrangement in lung adenocarcinomas in a routine practice: a French study. J Thorac Oncol. 2012;7(2):348-54.
11. Cappuzzo F, Marchetti A. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J Clin Oncol. 2009;27(10):1667-74.
12. Sequist LV, Waltman BA. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26.
ARTICLE INFO
DOI: 10.12699/jfvp.6.17.2015.4
Conflict of Interest
Non
Date of manuscript receiving
22/11/2014
Date of publication after correction
12/02/2015
Article citation
Ngo C, Duong-Quy S. Molecular abnormalities and target therapy for non-small cell lung cancers. J Func Vent Pulm 2015;17(6):4-9.



Copyright: jfvpulm.com
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}