
 English
English
 French
French
 Pubmed
Pubmed Google Scholar
Google Scholar Cross Ref
Cross Ref Visistor: 1776318
Visistor: 1776318La maladie de Kikuchi Fujimoto: A propos de deux cas
Kikuchi Fujimoto’s disease: A report of two cases
N. Amangar 1, H. Slimani 1, H.E.Ouazzani 2, K. Marc 1, M. Soualhi 1, R. Zahraoui 1, J. Benamor 1,
J.E. Bourkadi 1
1: Service de Pneumo-Phtisiologie. Hôpital Moulay Youssef. CHU Ibn Sina - Rabat. Maroc
2: Service de pneumologie. Hôpital Avicenne. CHU Ibn Sina - Rabat. Maroc
Corresponding author
Dr. Nadia AMANGAR
Service de Pneumo-Phtisiologie. Hôpital Moulay Youssef
CHU Ibn Sina - Rabat. Maroc
E-mail: amangar.nadia@gmail.com
ABSTRACT
Kikuchi-Fujimoto’s disease or histiocytic necrotizing lymphadenitis is a rare and benign disease predominantly occurring in young women, rarely the child, it is revealed by cervical lymphadenitis associated or no to systemic manifestations.
The diagnosis is based on the histological examination of a lymph node biopsy. Disease course is generally favourable with spontaneous resolution within few weeks. Kikuchi’sdisease can reveal or evolve into autoimmune disease particularly lupus, thus a long clinical and biological follow-up is necessary.
We report two cases of patients who developed Kikuchi disease: a pediatric case, and another case of a young adult.
KEYWORDS: Kikuchi-Fujimoto disease, histiocytic necrotizing lymphadenitis, lymphadenopathies
RÉSUMÉ
La maladie de Kikuchi-Fujimoto ou lymphadénite histiocytaire nécrosante est une affection rare et bénigne qui touche principalement la femme jeune, rarement l’enfant, elle est révélée par des adénopathies souvent cervicales associées ou non à des manifestations systémiques.
Le diagnostic repose sur l’histologie ganglionnaire, l’évolution est, généralement, favorable avec guérison spontanée au bout de quelques semaines. La maladie de Kikuchi peut révéler ou évoluer vers une maladie auto-immune, notamment lupique imposant un suivi clinicobiologique à long terme.
Nous rapportons deux observations de patients ayant développé une maladie de Kikuchi: un cas pédiatrique, et un autre cas d’un adulte jeune.
MOTS CLES: Maladie de Kikuchi-Fujimoto, lymphadénite histiocytaire nécrosante, adénopathies
INTRODUCTION
La maladie de Kikuchi - Fujimoto (MKF) ou lymphadénite histiocytaire nécrosante est une entité clinicopathologique distincte, rare et bénigne, d’étiologie inconnue, qui touche principalement les jeunes femmes avec prédilection en Asie. C’est une cause souvent méconnue d’adénopathies cervicales. Le diagnostic est avant tout histologique. Très peu de cas ont été rapportés chez l’enfant. Nous nous proposons de rappeler cette pathologie à partir de deux observations et des données de la littérature.
OBSERVATION N°1
Il s’agit d’un garçon de 11 ans, d’origine marocaine, sans antécédents pathologiques, hospitalisé pour exploration d’adénopathies cervicales bilatérales, associées à des myalgies, une fièvre à 38,5°C, des sueurs nocturnes, et une altération de l’état général évoluant depuis 3 semaines malgré un traitement antibiotique par une amoxicilline protégée.
L’examen clinique a retrouvé des adénopathies jugulaires bilatérales, centimétriques, mobiles, douloureuses à la palpation, et un œdème palpébral bilatéral. L’hémogramme a objectivé une leucopénie à 3 680/mm3, une lymphopénie à 1 200/mm3. La vitesse de sédimentation était accélérée à 70 mm la 1ère heure. La protéine C réactive était normale. Le taux des lacticodéshydrogénases était normal. Les sérologies de l’hépatite B et C, de la toxoplasmose, de la rubéole, du VIH, du cytomégalovirus et de l’Epstein Barr étaient négatives. La recherche de BK dans les crachats était négative. L’intradermoréaction à la tuberculine était à 10 mm.
La radiographie thoracique de face était normale.
L’échographie cervicale a confirmé la présence de multiples ganglions infra centimétriques à centre hypoéchogène, d’allure inflammatoire, siégeant au niveau jugulo-carotidien et spinal bilatéral plus marquées à gauche. L’échographie abdominale et le scanner thoracique était normaux.
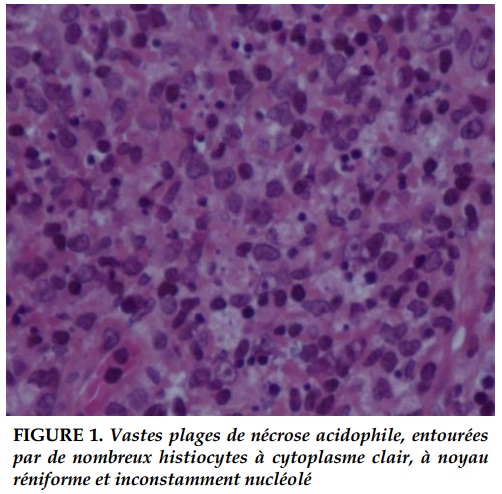
L’examen histologique de la biopsie ganglionnaire cervicale réalisée mettait en évidence un tissu ganglionnaire remanié par de vastes plages de nécrose acidophile non suppurées, entourées par une population cellulaire polymorphe comportant de nombreux histiocytes à cytoplasme clair, à noyau réniforme et inconstamment nucléolé. Cet aspect histologique était en faveur d'une maladie de KikuchiFujimoto (Figure 1).
Aucun traitement n’a été administré, et l’évolution a été spontanément favorable avec disparition des
adénopathies en quelques semaines.
OBSERVATION N°2
Il s’agit d’une patiente âgée de 30 ans, sans antécé-dents particuliers, qui a consulté pour une masse cervicale droite. L’examen clinique a retrouvé une patiente fébrile à 38°C, avec une adénopathie cervicale latérale droite, de 2 cm de diamètre, douloureuse à la palpation, sans signe inflammatoire en regard, sans hépatomégalie, ni splénomégalie, ni autres signes associés, la palpation des autres aires ganglionnaires était normale.
L’échographie cervicale a confirmé la nature ganglionnaire de la masse au dépend de la chaine jugulo carotidienne droite. La radiographie thoracique était sans anomalies. Le bilan biologique a montré un syndrome inflammatoire modéré avec une vitesse de sédimentation à 50 mm la première heure, la numé-ration de la formule sanguine a montré une leucopé-nie à 3 000 éléments/mm3, sans lymphopénie ou autres anomalies de la formule sanguine, l’intradermoréaction à la tuberculine était à 12mm, la recherche de BK dans les expectorations était négative à l’examen direct, la sérologie HIV est revenue négative.
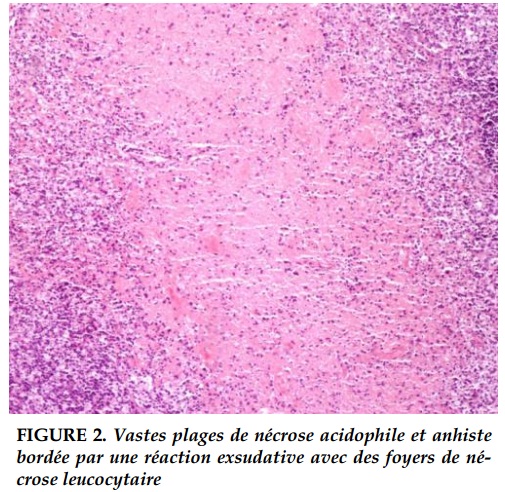
L’examen anatomopathologique de la biopsie ganglionnaire cervicale réalisée a retrouvé de vastes plages de nécrose acidophile et anhiste directement bordée par une réaction exsudative marquée, avec des foyers de nécrose leucocytaire concluant à une adénite nécrosante de Kikuchi (Figure 2).
Le bilan a été complété par la recherche d’anticorps anti nucléaire et anti DNA qui était négative, l’évolution spontanée était favorable après 6 mois.
DISCUSSION
La lymphadénite histiocytaire nécrosante est une maladie rare décrite en 1972 simultanément par 2 auteurs japonais, Kikuchi et Fujimoto, comme respectivement « une lymphadénite caractérisée par une prolifération focale de cellules réticulaires accompagnée de débris nucléaires et d’une phagocytose » et « une lymphadénite cervicale nécrosante subaiguë » [1].
C’est une affection de l’adulte jeune, avec une moyenne d’âge de 25 à 30 ans, et des extrêmes allant de un mois à 75 ans [2, 3]. Le sexe féminin prédomine dans presque toutes les séries, et varie de deux à neuf femmes pour un homme [4, 5]. Chez l’enfant [6], la particularité de cette affection est qu’elle inté-
resse les garçons préférentiellement d’origine africaine comme dans notre observation n°1. L’analyse des plus grandes séries de la littérature a permis de dé- gager les caractéristiques cliniques de cette maladie [7-9].
La lymphadénopathie représente l’anomalie clinique commune à tous les patients, en général isolée, mais qui peut être généralisée jusqu’à 20% des cas [4]. La localisation cervicale est la plus fréquente (88% des cas), le plus souvent au niveau de la chaine trapé- zienne ou jugulo-carotidienne [2, 4, 10] comme pour le cas des deux notre patients, mais toutes les chaines ganglionnaires peuvent être atteintes y compris chaines médiastinales et retropéritonéales [4]. Habituellement les ganglions de consistance variables parfois fermes, mobiles, non fluctuants, mesurant 1à 2cm, rarement plus de 3cm, ne s’ulcérant jamais [4, 11].
De rares cas d’adénopathies mesurant 10cm été rapportés [12]. Des manifestations générales peuvent être associées: une fièvre (37,4%), amaigrissement (7,5%), sueurs nocturnes (5,3%), une anorexie (7,5%) [4, 7-9]. Les localisations extranodales de la MKF sont peu fréquentes, variables, mais bien documentées ; l’atteinte cutanée est retrouvée dans 9,2% des cas [4, 7, 8, 13, 14], sous forme d’une variété de lésions non spécifiques, uniques ou multiples volontiers au niveau de la face, les deux membres supé-rieurs et la partie supérieure du tronc. On décrit des érythèmes parfois papuleux voire même pustuleux, des nodules, des plaques, une vascularite leucocytoclasique, et un syndrome de Wells [15], il peut aussi s’agir d’un œdème palpébral isolé comme le cas de notre patient ou de lésions en aille de papillon [7].
L’atteinte neurologique est rare au cours de la maladie, estimée à 5%, représentée essentiellement par une méningite lymphocytaire aséptique bénigne [16], les autres complications neurologiques [17] (ataxie cérébelleuse, atteinte du tronc cérébral, névrite optique, diplopie, hypertension intracrânienne, et encéphalomyélite aigue disséminée) sont moins fré- quentes, un cas de névrite optique révélant la maladie [30], et un autre cas d’état de mal épileptique inaugural ont été récemment décrits [31].
D’autres atteintes plus rares sont rapportées: des arthralgies [7, 13, 14], des myalgies [7, 13, 14], des douleurs thoraciques ou abdominales [18], une toux [7], des nausées -vomissements [7], une diarrhée [7], une odynophagie [14], une pharyngite, une parotidite [13], une hépatomégalie [14], une splénomégalies [14] , une néphromégalie [19], un trismus [13], des ulcérations des muqueuses [13], une épididymite [4], une rhabdomyolyse [19] , une choroidite [13], une uvéite antérieure bilatérale [13] .
Les signes biologiques sont dominés par un syndrome inflammatoire non spécifique [2, 14], une élévation des LDH [14], la leucopénie est retrouvée dans 49,2% des cas [11, 13], parfois on retrouve une neutropénie qui est souvent modérée associée à une lymphocytose relative [7], et exceptionnellement une bicytopénie ou pancytopénie [7]. Les anticorps antinucléaires sont habituellement négatifs, ils sont positifs en cas d’association à un lupus érythémateux disséminé ou à une autre connectivite [2, 11]. Une cytolyse hépatique, ou un syndrome d’activation macrophagique peuvent se rencontrer [2, 14, 20].
La confirmation diagnostique repose sur l’examen anatomopathologique d’une adénopathie qui permet aussi d’éliminer les principaux diagnostics différentiels: les lymphomes et les nécroses ganglionnaires infectieuses, notamment la tuberculose et la maladie des griffes du chat [21].
Les critères histopathologiques retenus par Kikuchi et al. sont [11]: des lésions focales bien circonscrites contenant un agrégat de larges lymphocytes, des histiocytes avec ou sans image de phagocytose et des débris nucléaires par caryorrhexis, principalement dans le paracortex et le cortex ganglionnaire, l’absence d’un nombre significatif de polynucléaires neutrophiles ou éosinophiles et de plasmocytes, et occasionnellement, une nécrose cellulaire importante, des dépôts de fibrine éosinophile, des histiocytes spumeux, prédominant autour des foyers de nécrose.
L’étude immunohistochimique montre la prédominance des cellules CD3+ et des histiocytes CD68+ dans les zones pathologiques [8]. Ainsi, la MKF peut être associée à d’autres maladies en particuliers auto immunes dont la liste continue à s’élargir: un lupus érythémateux disséminé [7, 8, 14, 21], parfois seulement un lupus cutané chronique [22], une maladie de Still [23], une polymyosite [24], une syndrome des anticorps antiphospholipides [25], une polyarthrite rhumatoïde [26], une sclérodermie[14], une connectivite indéterminée[27], une vascularite pulmonaire [20], un syndrome de Sjogren [20], un syndrome de Sharp[14], une thyroidite d’Hashimoto[14] ou cancer thyroïdiens [28], VIH [23], et d’autres cancers (estomac, sein, nasopharynx, poumon, ovaire) [7].
Des cas de MKF apparus au cours d’une grossesse ont été rapportés [29].
Généralement la maladie reste limitée et se résout spontanément en 1 à 4 mois, un traitement symptomatique (antipyrétique, antalgique, voir antiinflammatoire non stéroïdiens) est le plus souvent
suffisant ; lorsque les symptômes sont gênants (signes généraux importants, ganglions volumineux), la corticothérapie reste efficace [30]. Des rechutes peuvent survenir avec une moyenne de 3,7% [2, 7, 11, 13] nécessitant à chaque fois une confirmation histologique. Des cas mortels ont été rapportés dans la littérature, mais ne concernent que des patients ayant un système immunitaire défaillant [31].
L’apparition secondaire d’une pathologie autoimmune, notamment lupique nécessite un suivi prolongé [14].
CONCLUSION
La maladie de Kikuchi-Fujimoto est une pathologie bénigne qui doit être évoquée devant des adénopathies cervicales évoluant ou non dans un contexte fébrile. Le diagnostic est avant tout histologique. Les caractéristiques cliniques, biologiques et l’évolution sont identiques quel que soit l’âge. L’apparition secondaire d’une pathologie auto-immune, notamment lupique nécessite un suivi prolongé.
CONFLITS D’INTÉRÊTS
Aucun.
REFERENCES
1. Kikuchi M. Lymphadenitis showing focal reticulum cell hyperplasia with nuclear debris and phagocytosis. Nippon Ketsueki Gakkai Zasshi 1972; 35: 379-80.
2. Meyer O. La maladie de Kikuchi. Ann Med Interne 1999; 15: 199–204.
3. TsangWilliam YW, Chan JKC. Kikuchi’s lymphadenitis: a morphologic analysis of 75 cases with special reference to unusual features. Am J Surg Pathol 1994; 18: 219–31.
4. Turner RR, Martin J, Dorfman RF. Necrotizing lymphadenitis. A study of 30 cases. Am J Surg Pathol 1983; 7: 115–23.
5. Menasce LP, Banerjee SS, Edmondson D, Harris M. Histiocytic necrotizing lymphadenitis (KikuchiFujimoto disease): continuing diagnostic difficulties. Histopathology 1998; 3: 248–54.
6. Lin HS, Su CY, Huang SC. Kikuchi’s disease in asian children. Pediatrics 2005; 115: 92–6.
7. Kuo TT. Kikuchi’s disease (histiocytic necrotizing lymphadenitis): a clinicopathologic study of 79 cases with an analysis of histologic subtypes, immunohistology and DNA ploidy. Am J Surg Pathol 1995; 19: 798-809.
8. Lin HH et al. Clinical manifestations of Kikuchi’s disease in southern Taiwan. J Microbiol Immunol Infect 2005; 38: 35-40.
9. Lin HC, Su CY, Huang CC, Hwang CF, Chien CY. Kikuchi’s disease: a review and analysis of 61 cases. Otolaryngol Head Neck Surg 2003; 128: 650-3.
10. Raft J, Montinet B. Maladie de Kikuchi comme étiologie d’adénopathies cervicales chroniques. Rev Laryngol Otol Rhinol 2002; 123: 125–8.
11. Kikuchi M, Takeshita M, Eimoto T, Iwasaki H, Minamishima Y, Maedo Y. Histiocytic necrotizing lymphadenitis: clinicopathologic, immunologic and HLA typing study. In: Hanoaka M, Kadin ME, Mikata A, editors. Lymphoid malignancy: immunocytologic and cytogenetics. New York: Fields and Wood 1990. p. 251–7.
12. Unger PD, Rappaport KM, Strauchen JA. Necrotizing lymphadenitis (Kikuchi’s disease). Report of four cases of an unusual pseudolymphomatous lesion and immunologic marker studies. Arch Pathol Lab Med 1987; 111: 1031–4.

FIGURES
REFERENCES
1. Kikuchi M. Lymphadenitis showing focal reticulum cell hyperplasia with nuclear debris and phagocytosis. Nippon Ketsueki Gakkai Zasshi 1972; 35: 379-80.
2. Meyer O. La maladie de Kikuchi. Ann Med Interne 1999; 15: 199–204.
3. TsangWilliam YW, Chan JKC. Kikuchi’s lymphadenitis: a morphologic analysis of 75 cases with special reference to unusual features. Am J Surg Pathol 1994; 18: 219–31.
4. Turner RR, Martin J, Dorfman RF. Necrotizing lymphadenitis. A study of 30 cases. Am J Surg Pathol 1983; 7: 115–23.
5. Menasce LP, Banerjee SS, Edmondson D, Harris M. Histiocytic necrotizing lymphadenitis (KikuchiFujimoto disease): continuing diagnostic difficulties. Histopathology 1998; 3: 248–54.
6. Lin HS, Su CY, Huang SC. Kikuchi’s disease in asian children. Pediatrics 2005; 115: 92–6.
7. Kuo TT. Kikuchi’s disease (histiocytic necrotizing lymphadenitis): a clinicopathologic study of 79 cases with an analysis of histologic subtypes, immunohistology and DNA ploidy. Am J Surg Pathol 1995; 19: 798-809.
8. Lin HH et al. Clinical manifestations of Kikuchi’s disease in southern Taiwan. J Microbiol Immunol Infect 2005; 38: 35-40.
9. Lin HC, Su CY, Huang CC, Hwang CF, Chien CY. Kikuchi’s disease: a review and analysis of 61 cases. Otolaryngol Head Neck Surg 2003; 128: 650-3.
10. Raft J, Montinet B. Maladie de Kikuchi comme étiologie d’adénopathies cervicales chroniques. Rev Laryngol Otol Rhinol 2002; 123: 125–8.
11. Kikuchi M, Takeshita M, Eimoto T, Iwasaki H, Minamishima Y, Maedo Y. Histiocytic necrotizing lymphadenitis: clinicopathologic, immunologic and HLA typing study. In: Hanoaka M, Kadin ME, Mikata A, editors. Lymphoid malignancy: immunocytologic and cytogenetics. New York: Fields and Wood 1990. p. 251–7.
12. Unger PD, Rappaport KM, Strauchen JA. Necrotizing lymphadenitis (Kikuchi’s disease). Report of four cases of an unusual pseudolymphomatous lesion and immunologic marker studies. Arch Pathol Lab Med 1987; 111: 1031–4.
ARTICLE INFO
DOI: 10.12699/jfvp.5.15.2014.33
Conflict of Interest
Non
Date of manuscript receiving
12/01/2014
Date of publication after correction
17/8/2014
Article citation
Amangar N, Slimani H, Ouazzani H.E, Marc K, Soualhi M, Zahraoui R, Benamor J, Bourkadi J.E. Kikuchi Fujimoto’s disease: A report of two cases. J Func Vent Pulm 2014;05(15):33-37.



Copyright: jfvpulm.com