
 English
English
 French
French
 Pubmed
Pubmed Google Scholar
Google Scholar Cross Ref
Cross Ref Visistor: 1001
Visistor: 1001Effet de l’extrait de la fumée de cigarettes sur l’activité de Rhokinase, NOSe et ET-1 dans les cellules endothéliales pulmonaires
Effect of cigarette smoke on activity of Rho-kinase, eNOS, and ET-1 in endothelial pulmonary cells
Y. Bei1,2, S. Duong-Quy1,3
1: Université Paris Descartes. Faculté de Médecine Port Royal. UPRES 2511, Paris - France
2: Centre de Recherche Clinique et Translationnelle. Université de Médecine de Tongji, Shanghai, Chine
3: Collège de Médecine de Lam Dong. 6B Ngo Quyen, Dalat - Vietnam
Corresponding author
Dr. DUONG-QUY Sy
Collège de Médecine de Lam Dong - Dalat, Vietnam
Service de Physiologie - Explorations Fonctionnelles. Hôpital Cochin
E-mail: sy.duong-quy@cch.aphp.fr
ABSTRACT
Introduction. Cigarette smoke is associated with endothelial dysfunction and increased pulmonary vascular tone. However, changes in the RhoA/Rho-kinase/ROCK pathway related to the interaction between NO, ET-1, and cigarette smoke are not known.
Objectives. This work aims to study the effect of cigarette smoke extract (CSE) on the activity of RhoA/Rho kinase/ROCK signaling pathway and eNOS in endothelial cells (EC) of human pulmonary arteries.
Methods. In this study, the EC of human pulmonary arteries were stimulated with 3 different concentrations (1%, 2.5% and 5%) of CSE during 1h30 to 12h maximum. The protein expressions of ROCKs, endothelial NO synthase (eNOS), and ETB receptor were measured by Western blotting.
Results. After 12 hours of stimulation by CSE, the expression of ROCK-2 was obviously increased to 1% of CSE (0% vs 1%, 2520.70 ± 83.93 vs. 2863.78 ± 67.27, P <0.05). However, with 2.5% of the CSE, the expression of ROCK-2 has not continued to increase, but was significantly decreased compared to 1% of CSE (2.5% vs 1%, ± 2390.00 2863.78 ± 74.39 vs. 67.27, P <0.01). After 12 hours of stimulation, we found a significant decrease in the expression of eNOS in response to 5% of CSE (0% vs 5%, 540.06 ± 12.24 vs 436.30 ± 14.52, P <0.01).
Conclusion. In EC of pulmonary arteries, ROCK-2 might be activated by a low concentration of CSE but inhibited by a high concentration. The expression of eNOS might be inhibited by CSE of 5%.
KEYWORDS: Endothelial cells, pulmonary arteries, ROCK, eNOS, NO, cigarette smoke extract
RÉSUMÉ
Introduction. La fumée de cigarette est associée à la dysfonction endothéliale et à l’augmentation du tonus vasculaire pulmonaire. Cependant, les modifications de la voie RhoA/Rho-kinase/ROCK liées à l’interaction entre le NO, l’ET-1 et la fumée de cigarette ne sont pas connues.
Objectifs. Ce travail a pour objectif d’étudier l’effet de l’extrait de la fumé de cigarettes (EFC) sur l’activité de la voie de signalisation RhoA/Rho-kinase/ROCK et de NOSe dans les cellules endothéliales (CE) des artères pulmonaires humaines.
Méthode. Dans cette étude, les CE des artères pulmonaires humaines étaient stimulées par 3 concentrations (1%, 2.5% et 5%) d’EFC pendant 1h30 jusqu’à 12h maximum. Les expressions protéiques des ROCKs, de la NO synthase endothéliale (NOSe), des récepteurs ETB ont été mesurées par Western-Blot.
Résultats. Après 12 heures de stimulation par l’EFC, l’expression de la ROCK-2 était augmentée évidemment à 1% de l’EFC (0% vs 1%, 2520.70 ± 83.93 vs 2863.78 ± 67.27, P<0.05). Cependant, à 2.5% de l’EFC, l’expression de la ROCK-2 n’a pas continué à augmenter, mais était diminuée significativement par rapport à celle avec 1% de l’EFC (2.5% vs 1%, 2390.00 ± 74.39 vs 2863.78 ± 67.27, P<0.01). Après la stimulation de 12 heures, on a trouvé une diminution significative de l’expression d’NOSe sous l’effet de 5% de l’EFC (0% vs 5%, 540.06 ± 12.24 vs 436.30 ± 14.52, P<0.01).
Conclusion. Dans les CE vasculaires pulmonaires, la ROCK-2 peut être activée par de faibles concentrations de l’EFC, mais inhibée par de plus fortes concentrations et l’expression de la NOSe peut être inhibée par l’EFC à 5%.
INTRODUCTION
La consommation de tabac représente un problème majeur de santé publique avec des conséquences considérables sur le système respiratoire et cardiovasculaire, dont les plus communes sont la bronchopneumopathie chronique obstructive (BPCO), le cancer broncho-pulmonaire et l’athérosclérose. On estime que la consommation de tabac tue chaque année plus de 5 millions de personnes dans le monde. Si cette tendance persiste, le tabac pourrait causer chaque année plus de 8 millions de morts dans le monde d’ici à 2030 [1].
La fumée de cigarette contient plus de 4000 composants dont une majeure partie diffuse à travers la barrière alvéolo-capillaire et passe immédiatement dans la circulation pulmonaire. Au niveau des artères pulmonaires, les cellules endothéliales (CE) et les cellules musculaires lisses (CML) sont particulièrement sensibles aux effets nocifs de la fumée de cigarette. Les réponses anormales de ces cellules peuvent entraîner un dysfonctionnement endothélial, une augmentation du tonus vasculaire de base ainsi que des remaniements tissulaires des vaisseaux pulmonaires, qui pourraient ensuite conduire à une vasoréactivité pulmonaire anormale [2,3].
Au niveau du poumon, la voie de signalisation RhoA/Rho-kinase (ROCK) est impliquée dans la régulation de la vasoréactivité pulmonaire. La RhoA, une petite protéine G de la famille des Rho-GTPases couplée avec un récepteur membranaire, peut être activée non seulement par de nombreux médiateurs vasoactifs, mais aussi par les contraintes de cisaillement et l’hypoxie [4]. Les ROCKs (ROCK-1 et ROCK -2), effecteurs de la RhoA, ont de nombreux effets dans la régulation des fonctions cellulaires (migration, prolifération, apoptose), de l’expression génique et de la contraction des CML [5]. La modification de cette voie de signalisation joue un rôle important dans le développement de certaines maladies respiratoires associées à l’hypertension artérielle pulmonaire(HTAP) [6]. Si les effets de l’hypoxie sur l’activation de la voie RhoA/ROCK ont été déjà étudiés [7-9], peu d’études ont évalué les modifications de cette voie de signalisation sous l’effet du tabac.
De nombreux médiateurs vasoactifs dérivés de l’endothélium contrôlent la vasoréactivité pulmonaire. Le monoxyde d’azote (NO), un agent vasodilatateur, diminue le tonus vasculaire pulmonaire et inhibe la prolifération des CML [10]. A l’inverse, l’endothéline -1 (ET-1) est un puissant agent vasoconstricteur qui possède des effets mitogéniques sur les CML vasculaires [11]. De nombreuses études ont montré que la fumée de cigarette, et le stress oxydatif dû à la présence des ROS (reactive oxygen species), peut d’une part inhiber la synthèse et/ou l’activité du NO, et d’autre part stimuler la synthèse et/ou l’activité de l’ET-1, aboutissant à une augmentation du tonus vasculaire et un remodelage vasculaire pulmonaire [12].
L’effet du tabac sur la vasoréactivité pulmonaire est complexe, lié à l’existence de nombreuses interactions entre différentes voies de signalisation, telles que la voie NOSe/NO, la voie ET-1 et la voie RhoA/ ROCK. Par exemple, la voie RhoA/ROCK peut être activée par l’intermédiaire de l’ET-1 [13], alors qu’elle peut aussi être inhibée par les donneurs du NO [14]. Jusqu’à présent, bien que les effets de la fumée de cigarette sur la vasoréactivité pulmonaire par l’intermédiaire de la voie NOSe/NO et de la voie ET-1 soient connus, le rôle de la voie RhoA/ROCK dans ces interactions entre NO et ET-1, notamment lors de l’exposition aux fumées de tabac, reste à préciser.
OBJECTIFS
Cette étude a été réalisée pour étudier : 1) Les expressions des Rho-kinases (ROCK-1 et ROCK-2) et l’expression de la NOSe (endothelial NOS) et l’expression du récepteur de l’ET-1 (ETB) sous l’effet aigu de la fumée de cigarette ; 2) Chercher le mécanisme moléculaire lié aux interactions entre la voie RhoA/ROCK, la voie NOSe/NO et la voie ET-1 sous l’effet aigu de la fumée de cigarette.
MATERIELS ET METHODES
Culture cellulaire
Les matériels pour la culture cellulaire sont principalement fournis par Promocell sauf qu’on indique autre part. Les CEs (Human Pulmonary Artery Endothelial Cells, HPAEC, C-12241) des artères pulmonaires humaines sont décongelées et cultivées avec les milieux spéciaux (Endothelial Cell Growth Medium MV 2, C-22022 plus 100 IU/mL de pénicilline, 100 IU/mL de streptomycine et 0.25 ug/mL de fungizone, dans les flacons de 25 cm2 à 37°C avec 5% de CO2. Le 3ème au 7ème passage des CE et le 7ème au 11ème passage des CML sont utilisées dans tous nos expériences. Quand les cellules sont proliférées jusqu’à la confluence de 90%, elles sont stimulées avec différentes concentrations de l’EFC par rapport au contrôle.
Préparation de l’EFC
L’EFC a été préparé selon la méthode validée par David Bernhard et al [15] avec quelques modifications. En bref, 3 cigarettes commerciaux (CAMEL, 0.8 mg Nicotine, 10mg Goudrons) ont été bouffées dans 5 mL de milieu RPMI 1640 (7~8 bouffées/ cigarette, 20 bouffées/ 5 mL) sous la hotte. Les cigarettes ont été bouffées à l’aide d’une pompe automatique respiraroire (Smoking machine RM 1/ G borgwaldt, Germany) avec une fréquence de 1 bouffée/ min (chaque bouffée est durée pendant 2 secondes) , en fabriquant 35 mL de fumée/ bouffée, qui imite l’habitude des fumeurs [16]. L’EFC préparé préalablement, considéré comme la solution mère (100%), était ensuite conservé à -20°C jusqu’à l’utilisation, dont la qualité a été évaluée par la mesure de l’absorbance à 320 nm où se trouve la longueur d’onde specialisée pour le peroxynitrite[72]. Après une fois de dilution avec le milieu RPMI 1640 (V/V), l’EFC avec une absorbance de 3.0 ± 1.0 a été utilisé. Après, la solution mère (100%) a été diluée par volume de milieu spécialisé, avec une échelle de concentation de 1% jusqu’à 5% dans les expériences suivantes. Le 1% de l’EFC (0.04 bouffée/ mL) correspond à une consommation de 1 à 1.5 paquets de cigarettes par jour pour les fumeurs, chez qui la concentration plasmatique de nicotine est de 20-40 ng/ mL.
Test de cytotoxicite par MTT
Pour évaluer la cytotoxicité de l’EFC, la viabilité des cellules a été mesurée par le test au MTT (3-(4,5diméthylthiazol-2-yl)-2,5-diphényl tétrazolium bromide), qui est basé sur la transformation du MTT en cristaux bleus de formazan par une enzyme mitochondriale, la succinate déshydrogénase. Les cristaux de formazan formés sont solubilisés dans le DMSO et peuvent ainsi être mesurés par spectrophotométrie à une longueur d’onde de 550 nm.
En bref, les cellules étaient ensemencées dans une plaque de 96 puits, avec une confluence de 10,000 cellules dans 200 µL de milieu. Au moins 12 heures après l’ensemencement, les cellules étaient exposées aux différentes concentrations de l’EFC (0% à 30%) pendant 24h. Après 24 heures de stimulation, 50µL de 0.2% MTT (Sigma) a été ajouté dans chaque puits en jettant l’ancien milieu. Après 4 heures d’incubation avec MTT à 37°C, 100 µL/puits de DMSO a été ajouté au lieu de MTT. Finalement, l’absorbance a été mesurée à 550 nm, par un lecteur de plaques ELISA (Packard BioScience). Chaque concentration de l’EFC était réalisée en triple.
Stimulation des cellules par l’EFC
Une fois que des cellules sont proliférées à une confluence de 90%, elles sont exposées aux différentes concentrations de l’EFC (0%, 1%, 2.5% et 5%) pendant une courte durée. Les CE,réparties en deux groupes, sont exposées aux différentes concentrations de l’EFC pendant 3 h et 12 h, respectivement: groupe 1 : 3 h (EFC : 0%, 1%, 2.5% et 5%) ; groupe 2 : 12 h (EFC : 0%, 1%, 2.5% et 5%).
Western-Blot (WB)
Les homogénéités cellulaires ont été utilisées pour le WB après avoir été dosées avec le Pierce BCAProtein Assay Kit - Reducing Agent Compatible (﹟23250, Thermo Scientific) selon le protocole du fournisseur. En bref, 1 mL de solution BCA-A et 20 µL de solution BCA-B (50 :1), plus 25µL de l’homogénéité cellulaire étaient incubées ensemble dans un microtube de 1.5 mL à 37°C pendant 30 minutes. Après l’autozéro de la spectrophotométrie avec le RIPA 1X, la concentration totale des protéines a été mesurée à 562 nm, dont l’unité est µg/µL.
Preparation des echantillons
Selon la concentration totale des protéines mesurée, les échantillons pour Western-Blot ont été préparées avec Laemmli (4X), antiprotéase (5X) et de l’eau désionisée. Finallement, la quantité totale des protéines dans chaque échantillon était déjà homogénéisée, avec une concentration finale de 20µg/ 50µL. Une échantillon de 300 µl comprend 120µg de protéines, 75µl de Laemmli (4X) et 60µl de l’antiprotéase (5X), en étant complétée par l’eau désionisé. Les échantillons devaient être préchauffées à 96°C pendant 7 à 8 minutes pour dénaturer les proteines avant la mesure par Western-Blot.
Préparation du gel
Le gel SDS-PAGE (7.5%/4%) était utilisé pour la migration des protéines. Finallement, les protéines de grands poids moléculaires, comme ROCK-1(160 kDa), ROCK-2(160 kDa), et NOSe (140 kDa), se trouvaient dans la partie supérieur du gel. Alors que les protéines de faibles poids moléculaires, comme le ETB (35 kDa), se trouvaient dans la partie inférieur du gel.
Migration des protéines
Pour garantir les bons résultats, une certaine quantité de protéine totale (5 µg pour NOSe, 15 µg pour ROCK-1, ROCK-2 et ETB) a été déposée dans chaque puits. La migration a été réalisée dans le tampon de migration (Tris 50 mM, Glycine 0.38 M, SDS 0.1% et de l’eau désionisée) avec l’appareil de Bio Rad.
Transfert des protéines
La membrane polyvinylidene fluoride (PVDF, Immobilon-P) a été choisie pour le transfert semi-sec en utilisant l’appareil Trans-Blot SD (Bio Rad), selon le protocole recommandé par le fournisseur (Millipore Corporation, USA).
Incubation avec des anticorps spécifiques
Après le transfert, la membrane PVDF a été rincée 2 fois de 5 minutes dans la PBS-Tween (1%) et puis incubée dans la PBS-Tween (1%)-Lait écrémé (5%) pendant 1 heure pour bloquer les sites nonspécifiques. La membrane a été ensuite incubée avec le 1er anticorps dilué dans la même solution de blocking sur l’agitateur pendant la nuit à 4°C. La concentration du 1er anticorps était de 1/500 pour ROCK-1 (SC-6055, Santa cruz), 1/1000 pour ROCK-2 (SC1851, Santa cruz), ETB (SC-21196, Santa cruz) et NOSe (SC-654, Santa cruz). Le lendemain, après 3 rinçages de 10 minutes avec la PBS-Tween (1%), la membrane était incubée ensuite avec l’anticorps secondaire pendant 45 minutes à la température ambiante et était encore rincée pendant 3 fois avant la révélation des protéines.
Pour contrôler la quantité totale des protéines dans chaque puits, la protéine -actine a été mesurée (dilution 1/1500, SC-47778, Santa cruz) comme un contrôle intérieur après stripping. En bref, les anticorps sur la membrane ont été enlevés par le rinçage dans le tampon de stripping (62.5 mM Tris-Hcl, 2% SDS, 100 mM -mercapto-éthanol) à 56°C pendant 30 minutes. Puis, les mêmes étapes ont été réalisés comme ce qui sont citées ci-dessus pour WesternBlot à partir de blocking. En effet, l’expression de protéine étudiée est présentée en rapport avec actine.
Détection des complexes antigène-anticorps
Le kit ECL Western blotting detection (Amersham, GE Healthcare) était utilisé pour la détection luminescente grâce à sa haute sensibilité et sa détection rapide. La luminescence est l'émission de lumière résultant de l'énergie émise par une substance qui passe de l'état excité à l’état fondamental.
Le film Hyper-film (Amersham, GE Healthcare) a été choisi pour révéler les signaux des complexes antigène-anticorps. Le temps d’exposition était de 5 secondes à 10 minutes en fonction des images.
Analyse statistique
Les expressions des protéines ont été mesurées par le densitomètre et contrôlées avec -actine. Au moins 3 mesures ont été réalisées pour chaque groupe. Une comparaison par l’analyse de variance (ANOVA) et le test de T-student indépendant sont utilisés pour l’analyse statistique des résultats. Finalement, les résultats de Western-Blot sont présentés par les valeurs moyennes ± SEM. Une valeur de p0.05 est considérée comme significative.
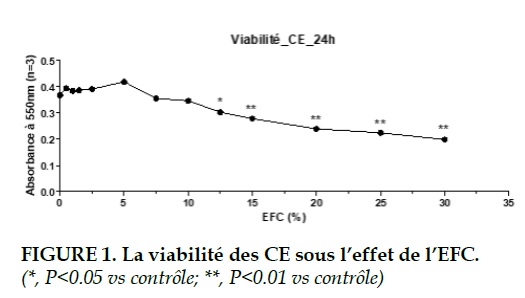
Cytotoxicite de l’EFC
Pour évaluer la cytotoxicité de l’EFC, la viabilité cellulaire a été mesurée initialement par le test au MTT. Après une stimulation par une échelle de 0% à 30% de l’EFC pendant 24 heures, une relation dose-effet a été montrée. Pour les CE, il n’y avait pas de différence significative sur la viabilité jusqu’à 10% de l’EFC.
Mais avec 12.5% de l’EFC, la viabilité des CE était diminuée signficativement par rapport au contrôle (P<0.05). La cytotoxicité était d’autant plus impor
Expression de la voie RhoA/ROCK dans les CE
Stimulation pendant 3 heures
Selon les résultats de la cytotoxicité de l’EFC, puisque les CE supportaient mieux l’EFC que les CML et qu’on n’a pas trouvé la différence significative sur les expressions des ROCKs dans les CML après 1h30 de stimulation par l’EFC, donc on a commencé avec 3 heures de stimulation pour les CE. Dans ce groupe-là, aucune différence significative a été trouvée sur les expressions des ROCKs.
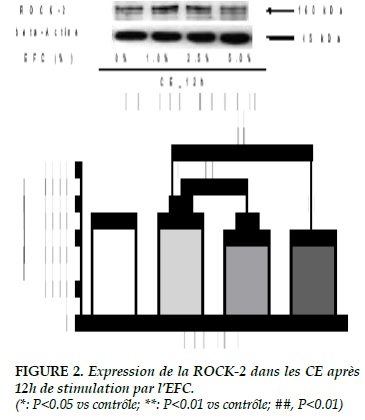
Stimulation pendant 12 heures
Tandis qu’après 12 heures de stimulation par l’EFC, l’expression de la ROCK-2 était augmentée évidemment à 1% de l’EFC (0% vs 1%, 2520.70 ± 83.93 vs 2863.78 ± 67.27, P<0.05). Cependant, à 2.5% de l’EFC, l’expression de la ROCK-2 n’a pas continué à augmenter, mais était diminuée significativement par rapport à celle avec 1% de l’EFC (2.5% vs 1%, 2390.00 ± 74.39 vs 2863.78 ± 67.27, P<0.01) De plus, son expression était encore diminuée énormément à 5% de l’EFC, même si par rapport au contrôle (5% vs 0%)
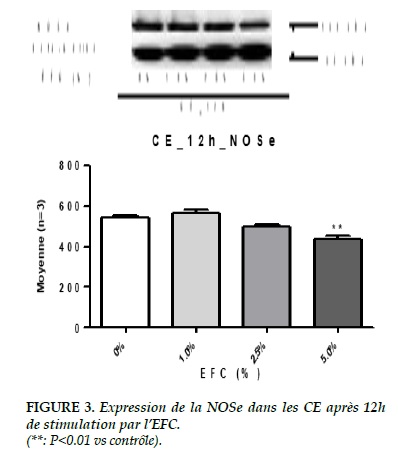
Expression de la NOSe dans les CE
Pour le groupe de 3h, il n’y avait pas de différence significative sur l’expression de la NOSe. Mais après la stimulation plus longue de 12 heures, on a trouvé une diminution significative de son expression sous l’effet de 5% de l’EFC (0% vs 5%, 540.06 ± 12.24 vs 436.30 ± 14.52, P<0.01) (Figure 3).
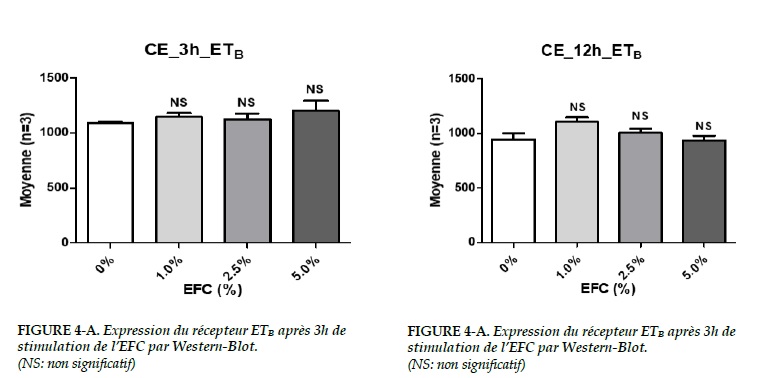
Récepteur ETB dans les CE
Dans les CE, aucune différence significative a été trouvée sur l’expression du récepteur ETB pour tous les deux groupes (3 h et 12 h) (Figure 4A-B).
DISCUSSION
Notre étude a montré que, dans les CE vasculaires pulmonaires soumises à des durées de stimulation plus longues, la protéine kinase ROCK-2 était activée par de faibles concentrations de l’EFC, tandis que son expression ainsi que l’expression de la NOSe étaient diminuées par de fortes concentrations de l’EFC.
La mesure de la cytotoxicité induite par l’EFC a été réalisée avec une échelle de concentration de l’EFC (0% à 30%) au début de l’étude avec le test au MTT. Les trois concentrations de l’EFC (1%, 2.5%, 5%), utilisées dans l’étude suivante, sont respectivement équivalentes aux concentrations considérées comme faible, modérée et forte par rapport aux concentrations plasmatiques de nicotine habituelles chez les fumeurs (43.7 ± 38 ng/mL) [17]. Ici, 1% de l’EFC (0.04 bouffée/mL) correspond à une consommation de 1 à 1,5 paquets de cigarettes pour les fumeurs, chez qui la concentration plasmatique de nicotine est de 20—40 ng/mL [15].
Bien que les résultats préliminaires ex vivo de notre laboratoire aient trouvé une activation de cette voie de signalisation au niveau des artères pulmonaires chez les fumeurs sans BPCO, le mécanisme moléculaire de cette activation n’est pas encore bien connu. Nous formulons l’hypothèse impliquant la voie ET1/ETA. Cette hypothèse est fondée sur un faisceau d’arguments que nous allons énumérer. L’ET-1 n’est pas stockée dans les vésicules de sécrétion au niveau des CE [18]. La production de l’ET-1 peut être régulée assez vite, en seulement quelques minutes, pour modifier le tonus vasculaire en réponse à certaines conditions comme l’hypoxie et l’ischémie [19]. La concentration plasmatique de l’ET-1 est plus élevée chez les fumeurs [20, 21]. Enfin, plusieurs études ont trouvé une augmentation de la production de l’ET-1 par les CE sous l’effet de l’EFC. Ainsi, Sang-Do Lee et al ont trouvé qu’une heure d’exposition des CE vasculaires pulmonaires entraînait une augmentation de l’expression génique de la pré-pro ET-1 alors que 4 h d’exposition augmentait de façon significative la production de l’ET-1 [22].
En dehors des résultats montrés ci-dessus, on a aussi trouvé qu’après 3h de stimulation avec une forte concentration de l’EFC (5%), l’expression de la voie RhoA/ROCK était significativement diminuée par rapport à celles obtenues avec 1% et 2.5% de l’EFC. En même temps, il y avait une importante diminution de l’expression du récepteur ETA par rapport à 1% et 2.5% de l’EFC, mais aussi par rapport aux témoins. Puisqu’on a déjà montré qu’il n’y avait pas de différence significative sur la mortalité des cellules à 5% de l’EFC, d’autres mécanismes sont donc mis en jeu pour expliquer l’inhibition de l’activation de la voie RhoA/ROCK avec de fortes concentrations de l’EFC.
Comme déjà indiqué ci-dessus, la fumée de cigarette contient de fortes quantités de NO pouvant interférer avec la voie RhoA/ROCK et la voie ET-1/ETA. Le NO est connu pour être capable d’inhiber l’activité de la RhoA par la S-nitrosation [23]. De plus, l’activité de la RhoA peut aussi être inhibée par la phosphorylation selon le niveau du NO [24]. Dans les CML vasculaires, le NO augmente la concentration de GMP cyclique (GMPc) en activant la guanylate cyclase soluble (GCs). Le GMPc active la protéine kinase G (PKG). La PKG participe ensuite à la phosphorylation de la RhoA, qui permet le GDI de capturer la RhoA phosphorylée dans le cytosol et finalement empêche son translocalisation à la membrane cellulaire où elle pourra être activée. Le NO est également capable de contrôler l’expression génique et protéique de la RhoA par l’activation de la PKG [25], et inhiber l’expression génique de la prépro ET-1 par l’intermédiaire de la GMPc dans les CE [26, 27].
Puisque les CE vasculaires pulmonaires supportaient mieux l’EFC selon les résultats du test de cytotoxicité, nous avions opté pour une durée d’exposition de 12h, au décours de laquelle l’expression de la ROCK2 était augmentée significativement par 1% de l’EFC. Il est déjà connu que l’activation de la voie RhoA/ ROCK dans les CE joue un rôle important dans le remaniement du cytosquelette et la modification de perméabilité endothéliale, qui entraîne finalement une altération de la barrière endothéliale [28, 29]. Nous avons ainsi montré que la voie RhoA/ROCK peut être partiellement activée dans les CE sous l’effet de faibles concentrations de l’EFC, ce qui pourrait altérer la barrière endothéliale et aggraver l’effet nocif du tabac sur des vaisseaux pulmonaires. A 5% de l’EFC, l’expression de la ROCK-2 était nettement diminuée par rapport au contrôle, ce qui pourrait être expliqué par le fait qu’une activation excessive de la voie NO/GMPc/PKG participe à la phosphorylation de la RhoA et inhibe son activité [24, 25].
En ce qui concerne la NOSe, nous n’avions trouvé une diminution de son expression qu’après 12 heures de stimulation par de fortes concentrations de l’EFC (5%). Plusieurs études ont montré que la fumée de cigarette était capable de diminuer la biosynthèse et la biodisponibilité du NO. Ainsi, Su et al ont trouvé que l’expression génique et protéique de la NOSe ainsi que son activité était diminuée de façon non réversible après la stimulation des CE vasculaires pulmonaires par l’EFC [30].
Zhang et al ont trouvé que l’activité de la NOSe était diminuée suite à une altération du métabolisme de l’Arginine causée par le tabac, et la production résultante des ROS [31]. De plus, Takemoto et al ont trouvé que l’expression de la NOSe dans les CE était diminuée suite à l’hypoxie et l’activation de la RhoA [32]. En fait, la voie RhoA/ROCK est capable d’inhiber non seulement l’expression mais aussi l’activité de la NOSe dans les CE [33, 34].
La modification de l’expression du récepteur ETB n’était pas significative dans les CE vasculaires pulmonaires. Le récepteur ETB, exprimé préférentiellement par les CE, contribue non seulement à la relaxation des CML en favorisant la production du NO et de la PGI2 par les CE, mais aussi à la clairance de l’ET-1 circulante [35]. Cependant, son rôle dans la modification de la voie RhoA/ROCK est très peu connu. Chez les fumeurs, même si l’inhalation d’une forte concentration de l’EFC peut entraîner transitoirement une vasodilatation pulmonaire grâce au NO, la diminution à long terme de la production basale du NO et les autres effets nocifs induits par la fumée de cigarette tels que l’altération de la fonction endothéliale, le remodelage vasculaire pulmonaire, finissent par entraîner des effets globalement nocifs.
CONCLUSIONS ET PERSPECTIVES
En conclusion, les résultats de notre travail ont permis de préciser que dans les CE vasculaires pulmonaires, après une stimulation relativement plus longtemps par l’EFC la ROCK-2 peut être activée par de faibles concentrations de l’EFC, mais inhibée par de plus fortes concentrations de l’EFC. De plus, l’expression de la NOSe peut être inhibée par l’EFC à 5%. Ces résultats nous incitent à penser que la voie ET-1/ETA pourrait être une cible importante si l’on voulait moduler l’effet du tabac sur l’activation de la voie la modification de cette voie en relation avec le NO reste encore à préciser. En sachant que le tabagisme est la première cause de mortalité évitable dans le monde, on espère que les recherches sur le mécanisme moléculaire de ses effets nocifs sur les vaisseaux pulmonaires nous permettront de mieux comprendre son rôle dans le développement et l'aggravation de certaines maladies telles que la BPCO et l'athéroscléose.
Remerciements. Les auteurs remercient le Dr T. HuaHuy et le Pr. AT. Dinh-Xuan pour leur aide dans la réalisation de cette étude.
CONFLIT D’INTERÊTS
Aucun.
REFERENCES
1. http://www5.who.int/tobacco/
2. Puranik, R., and Celermajer, D. S. Smoking and endothelial function. Prog. Cardiovasc. Dis. 2003;45:443–458.
3. S. Santos, V.I. Peinado, J. Ramírez, T. Melgosa, J. Roca, R. Rodriguez-Roisin, J.A. Barbera. Characterization of pulmonary vascular remodelling in smokers and patients with mild COPD. Eur Respir J 2002; 19: 632–638.
4. Barbara E. Rolfe, Nathalie F. Worth, Cameron J. World, Julie H. Campbell, Gordon R. Campbell. Rho and vascular disease. Atherosclerosis 2005;183:1–16.
5. Gervaise Loirand, Patrice Guérin and Pierre Pacaud. Rho Kinases in Cardiovascular Physiology and Pathophysiology. Circ Res. 2006;98:322-334.
6. Nicholas W. Morrell,et al. Cellular and Molecular Basis of Pulmonary Arterial Hypertension. Journal of the American College of Cardiology 2009;54:S20–31
7. Wang Z, Jin N, Ganguli S, Swartz DR, Li L, and Rhoades RA. Rho-kinase activation is involved in hypoxia-induced pulmonary vasoconstriction. Am J Respir Cell Mol Biol 2001; 25: 628–635.
8. Nagaoka T, Morio Y, Casanova N, Bauer N, Gebb S, McMurtry I, and Oka M. Rho/Rho kinase signaling mediates increased basal pulmonary vascular tone in chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol 2004; 287: L665–L672.
9. Fagan KA, Oka M, Bauer NR, et al Attenuation of acute hypoxic pulmonary vasoconstriction and hypoxic pulmonary hypertension in mice by inhibition of Rho-kinase. Am J Physiol Lung Cell Mol Physiol 2004; 287: L656–L664.
10. Dinh-Xuan AT, Higenbottam TW, Clelland CA, et al. Impairment of endothelium-dependent pulmonaryartery relaxation in chronic obstructive lung disease. N Engl J Med 1991; 324: 1539-47.
11. Karen A Fagan, Ivan F McMurtry and David M Rodman. Role of endothelin-1 in lung disease. Respir Res 2001; 2: 90–101.
12. Joanne L. Wright, Hsin Tai and Andrew Churg. Vasoactive mediators and pulmonary hypertension after cigarette smoke exposure in the guinea pig. J Appl Physiol 2006; 100: 672–678.
13. Nikki L. Jernigan, Benjimen R. Walker, and Thomas C. Resta. Reactive oxygen species mediate RhoA/Rho kinase-induced Ca2+ sensitization in pulmonary vascular smooth muscle following chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 2008; 295: L515–L529.
14. Sauzeau V, Le JH, Cario-Toumaniantz C, Smolenski A, Lohmann SM, Bertoglio J, et al. Cyclic GMPdependent protein kinase signaling pathway inhibits RhoA induced Ca2+ sensitization of contraction in vascular smooth muscle. J Biol Chem 2000; 275:21722–9.
15. David Bernhard, Christian W. Huck, Thomas Jakschitz, Gerald Pfister, Blair Henderson, Günther K. Bonn, Georg Wick. Development and evaluation of an in vitro model for the analysis of cigarette smoke effects on cultured cells and tissues. Journal of Pharmacological and Toxicological Methods 2004; 50: 45–51.
16. Djordjevic, M., Fan, J., Ferguson, S., and Hoffmann, D. Self-regulation of smoking intensity. Smoke yields of the low-nicotine, low-.tar. cigarettes. Carcinogenesis 1995; 16:2015-2021.
17. Papadoyannis, I. N., Samanidou, V. F., & Stefanidou, P. G. Clinical assay of nicotine and its metabolite, cotinine, in body fluids by HPLC following solid phase extraction. Journal of Liquid Chromatography and Related Technologies 2002; 25:2315–2335.
18. Nakamura S, Naruse M, Naruse K, Demura H, Uemura H. Immunocytochemical localization of endothelin in cultured bovine endothelial cells. Histochemistry 1990;94:475-7.
19. F. Brunner, H. Stessel, W.F. Graier, The effects of intracellular Ca2+ concentration and hypoxia on basal endothelin-1 secretion by cultured porcine aortic endothelial cells, Agents Actions 1995; Suppl. 45: 269-273.
20. Haak T, Jungmann E, Raab C, and Usadel KH. Elevated endothelin-1 levels after cigarette smoking. Metabolism 1994; 43: 267–269.
21. Goerre S, Staehli C, Shaw S, and Luscher TF. Effect of cigarette smoking and nicotine on plasma endothelin1 levels. J Cardiovasc Pharmacol 1995; 26: S236–S238.
22. Sang-Do Lee, Dong-Soon Lee, Yong-Gam Chun, et al. Cigarette smoke extract induces endothelin-1 via protein kinase C in pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol 2001; 281: L403– L411.
23. Zuckerbraun BS, Stoyanovsky DA, Sengupta R, Shapiro RA, Ozanich BA, Rao J, Barbato JE, Tzeng E. Nitric oxide-induced inhibition of smooth muscle cell proliferation involves S-nitrosation and inactivation of RhoA. Am J Physiol Cell Physiol 2007;292(2):C824-31.
24. Loirand G, Guilluy C, Pacaud P. Regulation of Rho proteins by phosphorylation in the cardiovascular system. Trends Cardiovasc Med 2006;16:199–204.
25. Sauzeau V, Rolli-Derkinderen M, Marionneau C, et al. RhoA expression is controlled by nitric oxide through cGMP-dependent protein kinase activation. J Biol Chem 2003; 278:9472–80.
26. Adrian P. L. Smith, Eric A. G. Demoncheaux, and Tim W. Higenbottam. Nitric Oxide Gas Decreases Endothelin-1 mRNA in Cultured Pulmonary Artery Endothelial Cells. NITRIC OXIDE: Biology and Chemistry 2002; 6( 2): 153–159.
27. Lisa K. Kelly, Stephen Wedgwood, Robin H. Steinhorn and Stephen M. Black. Nitric oxide decreases endothelin-1 secretion through the activation of soluble guanylate cyclase. Am J Physiol Lung Cell Mol Physiol 2004; 286: L984–L991.
28. Konstantin G. Birukov. Small GTPase in mechanosensitive regulation of endothelial barrier. Microvasculair Research 2009; 77: 46-52.
29. Birukova AA, Smurova K, Birukov KG, Usatyuk P, Liu F, Kaibuchi K, Ricks-Cord A, Natarajan V, Alieva I, Garcia JG, Verin AD. Microtubule disassembly induces cytoskeletal remodeling and lung vascular barrier dysfunction: role of Rho-dependent mechanisms. J Cell Physiol. 2004;201: 55–70.
30. Su, Y., W. Han, C. Giraldo, Y. De Li, and E. R. Block. Effect of cigarette smoke extract on nitric oxide synthase in pulmonary artery endothelial cells. Am. J. Respir. Cell Mol. Biol. 1998; 19:819–825.
31. Wei-Zheng Zhang, Kylie Venardos, Jaye Chin-Dusting and David M. Kaye. Adverse Effects of Cigarette Smoke on NO Bioavailability: Role of Arginine Metabolism and Oxidative Stress. Hypertension 2006;48;278285.
32. Takemoto M, Sun J, Hiroki J, et al. Rho-kinase mediates hypoxia-induced downregulation of endothelial nitric oxide synthase. Circulation 2002; 106: 57–62.
33. Yoshiyuki Rikitake and James K. Liao. Rho GTPases, Statins, and Nitric Oxide. Circ Res 2005;97;1232-1235.
34. Duong-Quy S, Dao P, Hua-Huy T, Guilluy C, Pacaud P, Dinh-Xuan AT. Increased Rho-kinase expression and activity and pulmonary endothelial dysfunction in smokers with normal lung function. Eur Respir J. 2011;37:349-55.
35. Michael J. Boscoe, Andrew T. Goodwin, Mohamed Amrani, Magdi H. Yacoub. Endothelins and the lung. The International Journal of Biochemistry & Cell Biology 2000; 32: 41-62.

REFERENCES
1. http://www5.who.int/tobacco/
2. Puranik, R., and Celermajer, D. S. Smoking and endothelial function. Prog. Cardiovasc. Dis. 2003;45:443–458.
3. S. Santos, V.I. Peinado, J. Ramírez, T. Melgosa, J. Roca, R. Rodriguez-Roisin, J.A. Barbera. Characterization of pulmonary vascular remodelling in smokers and patients with mild COPD. Eur Respir J 2002; 19: 632–638.
4. Barbara E. Rolfe, Nathalie F. Worth, Cameron J. World, Julie H. Campbell, Gordon R. Campbell. Rho and vascular disease. Atherosclerosis 2005;183:1–16.
5. Gervaise Loirand, Patrice Guérin and Pierre Pacaud. Rho Kinases in Cardiovascular Physiology and Pathophysiology. Circ Res. 2006;98:322-334.
6. Nicholas W. Morrell,et al. Cellular and Molecular Basis of Pulmonary Arterial Hypertension. Journal of the American College of Cardiology 2009;54:S20–31
7. Wang Z, Jin N, Ganguli S, Swartz DR, Li L, and Rhoades RA. Rho-kinase activation is involved in hypoxia-induced pulmonary vasoconstriction. Am J Respir Cell Mol Biol 2001; 25: 628–635.
8. Nagaoka T, Morio Y, Casanova N, Bauer N, Gebb S, McMurtry I, and Oka M. Rho/Rho kinase signaling mediates increased basal pulmonary vascular tone in chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol 2004; 287: L665–L672.
9. Fagan KA, Oka M, Bauer NR, et al Attenuation of acute hypoxic pulmonary vasoconstriction and hypoxic pulmonary hypertension in mice by inhibition of Rho-kinase. Am J Physiol Lung Cell Mol Physiol 2004; 287: L656–L664.
10. Dinh-Xuan AT, Higenbottam TW, Clelland CA, et al. Impairment of endothelium-dependent pulmonaryartery relaxation in chronic obstructive lung disease. N Engl J Med 1991; 324: 1539-47.
11. Karen A Fagan, Ivan F McMurtry and David M Rodman. Role of endothelin-1 in lung disease. Respir Res 2001; 2: 90–101.
12. Joanne L. Wright, Hsin Tai and Andrew Churg. Vasoactive mediators and pulmonary hypertension after cigarette smoke exposure in the guinea pig. J Appl Physiol 2006; 100: 672–678.
13. Nikki L. Jernigan, Benjimen R. Walker, and Thomas C. Resta. Reactive oxygen species mediate RhoA/Rho kinase-induced Ca2+ sensitization in pulmonary vascular smooth muscle following chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 2008; 295: L515–L529.
14. Sauzeau V, Le JH, Cario-Toumaniantz C, Smolenski A, Lohmann SM, Bertoglio J, et al. Cyclic GMPdependent protein kinase signaling pathway inhibits RhoA induced Ca2+ sensitization of contraction in vascular smooth muscle. J Biol Chem 2000; 275:21722–9.
15. David Bernhard, Christian W. Huck, Thomas Jakschitz, Gerald Pfister, Blair Henderson, Günther K. Bonn, Georg Wick. Development and evaluation of an in vitro model for the analysis of cigarette smoke effects on cultured cells and tissues. Journal of Pharmacological and Toxicological Methods 2004; 50: 45–51.
16. Djordjevic, M., Fan, J., Ferguson, S., and Hoffmann, D. Self-regulation of smoking intensity. Smoke yields of the low-nicotine, low-.tar. cigarettes. Carcinogenesis 1995; 16:2015-2021.
17. Papadoyannis, I. N., Samanidou, V. F., & Stefanidou, P. G. Clinical assay of nicotine and its metabolite, cotinine, in body fluids by HPLC following solid phase extraction. Journal of Liquid Chromatography and Related Technologies 2002; 25:2315–2335.
18. Nakamura S, Naruse M, Naruse K, Demura H, Uemura H. Immunocytochemical localization of endothelin in cultured bovine endothelial cells. Histochemistry 1990;94:475-7.
19. F. Brunner, H. Stessel, W.F. Graier, The effects of intracellular Ca2+ concentration and hypoxia on basal endothelin-1 secretion by cultured porcine aortic endothelial cells, Agents Actions 1995; Suppl. 45: 269-273.
20. Haak T, Jungmann E, Raab C, and Usadel KH. Elevated endothelin-1 levels after cigarette smoking. Metabolism 1994; 43: 267–269.
21. Goerre S, Staehli C, Shaw S, and Luscher TF. Effect of cigarette smoking and nicotine on plasma endothelin1 levels. J Cardiovasc Pharmacol 1995; 26: S236–S238.
22. Sang-Do Lee, Dong-Soon Lee, Yong-Gam Chun, et al. Cigarette smoke extract induces endothelin-1 via protein kinase C in pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol 2001; 281: L403– L411.
23. Zuckerbraun BS, Stoyanovsky DA, Sengupta R, Shapiro RA, Ozanich BA, Rao J, Barbato JE, Tzeng E. Nitric oxide-induced inhibition of smooth muscle cell proliferation involves S-nitrosation and inactivation of RhoA. Am J Physiol Cell Physiol 2007;292(2):C824-31.
24. Loirand G, Guilluy C, Pacaud P. Regulation of Rho proteins by phosphorylation in the cardiovascular system. Trends Cardiovasc Med 2006;16:199–204.
25. Sauzeau V, Rolli-Derkinderen M, Marionneau C, et al. RhoA expression is controlled by nitric oxide through cGMP-dependent protein kinase activation. J Biol Chem 2003; 278:9472–80.
26. Adrian P. L. Smith, Eric A. G. Demoncheaux, and Tim W. Higenbottam. Nitric Oxide Gas Decreases Endothelin-1 mRNA in Cultured Pulmonary Artery Endothelial Cells. NITRIC OXIDE: Biology and Chemistry 2002; 6( 2): 153–159.
27. Lisa K. Kelly, Stephen Wedgwood, Robin H. Steinhorn and Stephen M. Black. Nitric oxide decreases endothelin-1 secretion through the activation of soluble guanylate cyclase. Am J Physiol Lung Cell Mol Physiol 2004; 286: L984–L991.
28. Konstantin G. Birukov. Small GTPase in mechanosensitive regulation of endothelial barrier. Microvasculair Research 2009; 77: 46-52.
29. Birukova AA, Smurova K, Birukov KG, Usatyuk P, Liu F, Kaibuchi K, Ricks-Cord A, Natarajan V, Alieva I, Garcia JG, Verin AD. Microtubule disassembly induces cytoskeletal remodeling and lung vascular barrier dysfunction: role of Rho-dependent mechanisms. J Cell Physiol. 2004;201: 55–70.
30. Su, Y., W. Han, C. Giraldo, Y. De Li, and E. R. Block. Effect of cigarette smoke extract on nitric oxide synthase in pulmonary artery endothelial cells. Am. J. Respir. Cell Mol. Biol. 1998; 19:819–825.
31. Wei-Zheng Zhang, Kylie Venardos, Jaye Chin-Dusting and David M. Kaye. Adverse Effects of Cigarette Smoke on NO Bioavailability: Role of Arginine Metabolism and Oxidative Stress. Hypertension 2006;48;278285.
32. Takemoto M, Sun J, Hiroki J, et al. Rho-kinase mediates hypoxia-induced downregulation of endothelial nitric oxide synthase. Circulation 2002; 106: 57–62.
33. Yoshiyuki Rikitake and James K. Liao. Rho GTPases, Statins, and Nitric Oxide. Circ Res 2005;97;1232-1235.
34. Duong-Quy S, Dao P, Hua-Huy T, Guilluy C, Pacaud P, Dinh-Xuan AT. Increased Rho-kinase expression and activity and pulmonary endothelial dysfunction in smokers with normal lung function. Eur Respir J. 2011;37:349-55.
35. Michael J. Boscoe, Andrew T. Goodwin, Mohamed Amrani, Magdi H. Yacoub. Endothelins and the lung. The International Journal of Biochemistry & Cell Biology 2000; 32: 41-62.
ARTICLE INFO
DOI: 10.12699/jfvp.3.9.2012.32
Conflict of Interest
Non
Date of manuscript receiving
15/3/2012
Date of publication after correction
15/10/2012
Article citation
Bei Y, Duong-Quy S. Effect of cigarette smoke on activity of Rho-kinase, eNOS, and ET-1 in endothelial pulmonary cells. J Func Vent Pulm 2012;03(09):32-39.



Copyright: jfvpulm.com