
 English
English
 French
French
 Pubmed
Pubmed Google Scholar
Google Scholar Cross Ref
Cross Ref Visistor: 804
Visistor: 804Pneumopathie interstitielle fibrosante liée à la sclérodermie systémique
Interstitial lung disease due to systemic sclerosis
S. Rivière
Service de Physiologie-Explorations Fonctionnelles Cardio-Respiratoires. Hôpital Cochin - Paris
Laboratoire Biologie - Physiologie des Maladies Respiratoires. UPRES - EA 2511. Université Paris Descartes
Corresponding author
Dr Sébastien RIVIERE
Service de Physiologie - EFR. Hôpital Cochin. 75014 Paris
E-mail: sebastien-riviere1@laposte.net
ABSTRACT
Interstitial lung disease (ILD) is a common manifestation of systemic sclerosis (SSc), mostly in diffuse forms of the disease. Even though ILD is severe in about 16% of patients, it is currently the main cause of mortality in SSc.
Pathophysiology is complex and not fully understood. SSc-related ILD corresponds to Nonspecific Interstitial Pneumonia (NSIP) in most cases, whereas Usual Interstitial Pneumonia (UIP) is less frequently encountered. Pulmonary function tests and high-resolution computerized tomography allow diagnosis, frequently before the occurrence of clinical symptoms, and should be regularly performed. Moreover, they provide prognostic information.
Treatment of ILD is not well established. It is currently based on immunosuppressive therapies. Cyclophosphamide, the most used drug, failed to prove clinical relevant benefits. Open studies reported mycophenolate mofetyl, azathioprine or rituximab in association or as alternatives to cyclophosphamide. Tyrosine kinase inhibitors are being evaluated.
Several authors proposed intensive immunosuppression followed by autologus hematopoietic stem cell transplantation in severe forms of SSc. Lung transplantation can be proposed in selected patients.
KEYWORDS: Systemic sclerosis, interstitial lung disease, prognosis, treatment
RÉSUMÉ
La pneumopathie interstitielle fibrosante (PIF) est une atteinte fréquente de la sclérodermie systémique (ScS), principalement dans les formes diffuses de la maladie. Même si elle n’est sévère que chez environ 16% des patients, elle représente aujourd’hui la première cause de mortalité dans la ScS. La physiopathologie est complexe et encore en partie méconnue. La pneumopathie interstitielle non-spécifique (Nonspecific Interstitial Pneumonia, NSIP) est la forme la plus fréquemment rencontrée, la pneumopathie interstitielle commune (Usual Interstitial Pneumonia, UIP) étant plus rare.
Les explorations fonctionnelles respiratoires et le scanner thoracique permettent le diagnostic, le plus souvent avant l’ apparition des signes cliniques et doivent donc faire partie de la surveillance régulière des patients. Ils apportent également des informations pronostiques.
Le traitement de la PIF n’est pas clairement établi. Il est actuellement basé sur les immunosuppresseurs. Le cyclophosphamide, qui est le plus utilisé, n’a pas montré de bénéfices cliniquement significatifs. Des études ouvertes ont rapporté l’ utilisation du mycophénolate mofetyl, de l’azathioprine ou du rituximab comme alternative ou en association au cyclophosphamide. Des études utilisant des inhibiteurs de tyrosine-kinases sont en cours.
Dans les formes sévères, certaines équipes proposent une intensification de l’immunosuppression suivie d’autogreffe de cellules souches hématopoïétiques. Enfin, la transplantation pulmonaire peut être envisagée chez certains patients.
MOTS CLES: Sclérodermie systémique, pneumopathie interstitielle, pronostic, traitement
INTRODUCTION
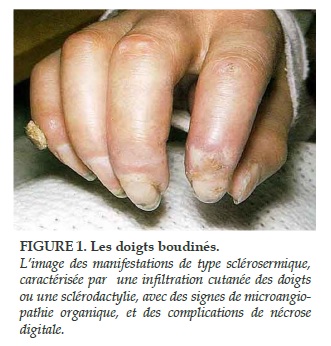
La sclérodermie systémique (ScS) est une pathologie caractérisée par une dysfonction endothéliale, des anomalies de régulation des fibroblastes à l’origine d’un excès de fabrication de collagène et des manifestions d’auto-immunité [1, 2]. Ces anomalies sont à l’origine d’une fibrose cutanée (Figure 1) et des organes internes, pouvant aboutir à des défaillances viscérales et au décès [3].
La maladie touche beaucoup plus fréquemment les femmes (ratio de 3 à 8 femmes pour 1 homme selon les séries). Sa prévalence est variable selon les régions, avec 276 par million d’habitants en Amérique du Nord [4], et 154 par million d’habitants en Europe [5].
Les atteintes viscérales les plus fréquentes sont pulmonaires, rénales, digestives et cardiaques. Le pronostic vital et fonctionnel des patients, autrefois principalement lié à la survenue de crises rénales sclérodermiques, dépend surtout aujourd’hui de l’existence d’une atteinte pulmonaire [6]. Les deux composantes majeures des manifestations pulmonaires de la ScS sont l’atteinte vasculaire d’ une part, avec la survenue d’une hypertension pulmonaire (HTP), et parenchymateuse d’autre part, liée à la pneumopathie interstitielle fibrosante (PIF).
Si l’amélioration des connaissances et l’arrivée de nouvelles thérapeutiques de l’HTP ont permis d’en améliorer la prise en charge, la physiopathologie de la PIF reste incomplètement comprise et les traitements actuels ont une efficacité limitée. L’objectif de cette revue est de développer les connaissances actuelles sur la PIF liée à la ScS en abordant notamment les aspects physiopathologiques et les perspectives thérapeutiques qui en découlent.
EPIDEMIOLOGIE ET CLASSIFICATION
La PIF est une des atteintes viscérales les plus fréquentes de la ScS, et touche beaucoup plus souvent les patients présentant une forme diffuse de la maladie. Sa prévalence varie selon les séries et les méthodes utilisées pour le diagnostic. Elle touche environ 40% des patients, mais des séries autopsiques ont retrouvé jusqu’à 80 à 100% d’atteinte du parenchyme pulmonaire [7]. Les formes sévères peuvent toucher jusqu’à 16% des patients présentant une forme diffuse de ScS [3].
La PIF est aujourd’hui la première cause de mortalité, représentant 33% des décès liés à la ScS et 16% de la mortalité globale [6]. Dans les formes sévères, elle diminue à 38% la survie à 10 ans [3, 6] alors que la survie globale à 10 ans des patients atteints de ScS varie entre 68% et 76,8% dans les séries récentes [6, 8].
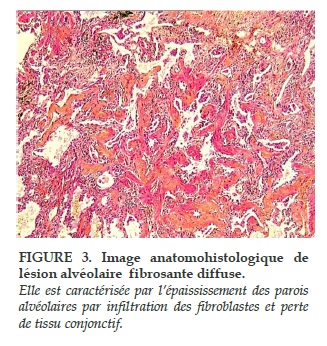
Un consensus des sociétés américaine et européenne de pathologie respiratoire a redéfini en 2002 la classification histologique des pneumopathies interstitielles [9]. La pneumopathie intersticielle non-spécifique (Nonspecific Interstitial Pneumonia, NSIP) est la forme la plus fréquemment rencontrée dans la ScS, devant la pneumopathie interstitielle commune (Usual Interstitial Pneumonia, UIP). Dans une série de biopsies pulmonaires sur 80 patients, leurs fréquences respectives étaient de 77,5% et 7,5% [10]. Beaucoup plus rarement des dommages alvéolaires diffus (Diffuse Alveolar Damage, DAD) et des pneumopathies organisées (Organizing Pneumonia, OP) peuvent être retrouvés [10-12]. Dans leur série de 80 patients, Bouros et al. n’ont pas montré de relation entre le type d’atteinte histologique et le pronostic [10].
PHYSIOPATHOLOGIE
La physiopathologie de la PIF est complexe et en grande partie méconnue à ce jour. Elle résulte probablement d’interactions anormales entre les cellules endothéliales, les cellules mononuclées (lymphocytes B et T, monocytes) et les fibroblastes [2]. Ces anomalies entraineraient le changement de phénotype des fibroblastes et l’excès de production de matrice extracellulaire (MEC) qui caractérise la fibrose. Nous ne développerons pas ici tous les aspects physiopathologiques, mais nous insisterons sur les mécanismes ayant à ce jour débouché sur des perspectives thérapeutiques.
Les anomalies endothéliales et l’infiltrat de cellules inflammatoires sont des phénomènes précoces [2, 13, 14]. Les lymphocytes Th2 sécrètent des cytokines qui favorisent la synthèse de MEC par les fibroblastes, en particuliers l’interleukine 4. De nombreuses autres cytokines sont impliquées, telles que le Transforming Growth Factor β (TGF β), le Connective Tissue Growth Factor (CTGF), le Platelet-Derived Growth Factor (PDGF) ou l’endothéline 1 [2, 15].
Les fibroblastes sont des cellules clés de la physiopathologie. Dans la ScS, ces cellules présentent de nombreuses différences par rapport aux fibroblastes normaux: sensibilité diminuée aux cytokines inhibant la synthèse de MEC [16] comme l’interféron γ, production accrue de cytokines comme le TGFβ, le CTGF, l’endothéline 1, anomalies des voies de signalisation intra-cellulaire du TGFβ [15, 17]. Ces anomalies soulignent le rôle d’une boucle autocrine amplifiant les phénomènes de fibrose.
Le rôle du stress oxydatif est important [2]. Les radicaux libres de l’oxygène sont produits en excès par plusieurs types cellulaires. Ils favorisent la production des facteurs pro-fibrosants et favorisent la production de collagène par les fibroblastes.
Enfin, l’implication du reflux gastro-œsophagien (RGO) dans la genèse de la PIF, par les micro-inhalations répétées, est débattue car plusieurs équipes ont rapporté des résultats contradictoires [18-20]. Dans une étude prospective avec un suivi de 2 ans, Marie et al. [19] ont montré qu’une atteinte œsophagienne sévère est associée à la dégradation de la capacité de diffusion du monoxyde de carbone (DLCO) et à des signes de PIF au scanner thoracique. Cette association ne permet cependant pas d’établir une relation de causalité et pourrait être le signe d’ une forme sévère de ScS, les atteintes œsophagiennes et pulmonaires étant fréquentes dans cette pathologie.
DIAGNOSTIC CLINIQUE
L’atteinte interstitielle pulmonaire est initialement asymptomatique et l’apparition d’une symptomatologie est souvent le reflet d’une forme évoluée. Les signes fonctionnels sont aspécifiques.
Les plus fréquents sont une dyspnée survenant principalement à l’effort, une toux sèche, une asthénie importante. Des douleurs thoraciques ou des hémoptysies sont rares et doivent faire rechercher d’éventuelles complications. L’examen physique peut retrouver des crépitants « velcro » des bases à l’auscultation pulmonaires, qui peuvent être présents avant l’apparition des signes fonctionnels. Des signes d’insuffisance cardiaque droite pouvant évoquer une hypertension pulmonaire associée doivent être recherchés.
EXAMENS PARACLINIQUES
Auto-anticorps
La majorité des patients présentent des anticorps anti-nucléaires. Les anticorps anti-topoisomérase I (initialement nommés anti-Scl 70) sont retrouvés chez 20 à 40% des patients, plus fréquemment dans les formes cutanées diffuses [21].
Explorations fonctionnelles respiratoires
La reproductibilité et le caractère non invasif des explorations fonctionnelles respiratoires font qu’elles ont une place majeure dans l’évaluation initiale et le suivi des patients. Elles comprennent les mesures des débits et volumes pulmonaires ainsi que de la capacité de diffusion du monoxyde de carbone (DLCO).
La PIF peut se manifester par un trouble de la diffusion alvéolo-capillaire (diminution de la DLCO) qui est souvent le signe le plus précoce [22] ou un trouble ventilatoire restrictif, défini par une diminution de la capacité pulmonaire totale (CPT). Beaucoup d’études ont utilisé la capacité vitale forcée (CVF) comme marqueur d’un trouble restrictif. Une diminution précoce de la CVF est associée à un risque de progression rapide de la PIF [23, 24] et à une mortalité plus élevée [10]. Une diminution de la DLCO est un marqueur de sévérité de la PIF [22] et est également associée à une mortalité plus élevée [10]. Toutefois une diminution isolée de la DLCO n’est pas spécifique de la PIF et peut être liée à une hypertension pulmonaire.
Le test de marche de 6 minutes avec monitorage de la saturation artérielle en oxygène et évaluation de la dyspnée, dont l’utilité a été démontrée dans le cadre de l’hypertension artérielle idiopathique, fait aujourd’hui partie des examens d’évaluation et de suivi des patients atteints de sclérodermie. Cependant, de nombreux facteurs confondants limitent sa spécificité, comme les atteintes articulaires et/ou musculaires qui sont fréquemment présentes au cours de la ScS [25, 26]. Son interprétation doit donc être prudente dans ce contexte.
Scanner thoracique haute résolution (TDM-HR)
Cet examen est beaucoup plus sensible que la radiographie du thorax. Schurawitzki et al. [27] ont retrouvé des signes d’atteinte interstitielle chez 91% des patients atteints de ScS alors que seuls 31% avaient des anomalies à la radiographie standard.
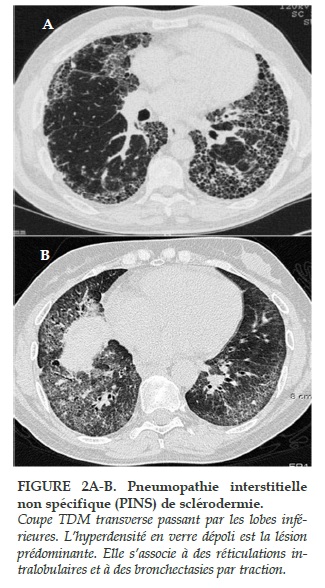
Au stade précoce de la PIF, des micronodules, des opacités en verre dépoli sont observés, avec une prédominance aux segments postérieurs des lobes inférieurs. Dans les formes plus évoluées apparaissent des opacités en rayon de miel et des bronchectasies par traction (Figure 2A et B). Il est probable que les opacités en verre dépoli soient les premiers signes de fibrose pulmonaire. En effet, une étude sur 41 patients ayant eu plusieurs TDM-HR consécutifs n’a montré une amélioration de ces lésions que dans 5% des cas alors que la majorité des patients étaient traités [28].
La TDM-HR apporte des informations pronostiques. Le degré d’extension des lésions est associé à une altération plus rapide des EFR et à une mortalité plus élevée [29]. A l’inverse, l’absence de lésions au moment du diagnostic est un critère de bon pronostic: dans une étude sur 90 patients, Launay et al. ont montré que 85% des patients ayant un scanner initial normal n’avaient pas développé de PIF pour une durée moyenne de suivi de 5 ans [30].
Lavage broncho-alvéolaire et biopsie pulmonaire
L’intérêt du lavage broncho-alvéolaire (LBA) chez les patients atteints de ScS a fait l’objet de très nombreuses études, dont les résultats ne sont pas concordants. L’existence d’une alvéolite (définie par une cellularité élevée) est retrouvée chez 38% des patients présentant des opacités en verre dépoli au scanner thoracique [31].
Elle est associée à une altération plus importante des explorations fonctionnelles respiratoires lors du diagnostic [32]. Une augmentation des polynucléaires neutrophiles (PNN) dans le LBA pourrait être associée à une mortalité précoce accrue [33]. Les données concernant l’élévation des polynucléaires éosinophiles (PNE) sont contradictoires [10, 33].
Dans une étude sur plus de 130 patients, Goh et al. ont montré que l’élévation des PNN, des PNE ou des lymphocytes n’étaient associées ni à la mortalité, ni à la survie sans progression, ni à une dégradation plus rapide des paramètres fonctionnels respiratoires [33].
De plus, dans une analyse sur les patients inclus dans la Scleroderma Lung Study [32], il n’a pas été retrouvé de lien entre l’existence d’une alvéolite et la réponse au traitement immunosuppresseur. L’ensemble de ces données, ainsi que le caractère invasif de cette procédure, font que la majorité des équipes réservent à l’heure actuelle le LBA à des situations de doute diagnostique comme la suspicion d’une infection respiratoire, chez des patients pouvant être immunodéprimés.
TRAITEMENT
Les options thérapeutiques pour cette atteinte grave de la ScS sont actuellement limitées. L’hypothèse d’un phénomène inflammatoire précoce dans la physiopathologie de la PIF a conduit à l’utilisation de traitements immunosuppresseurs, mais peu d’ études prospectives randomisées ont été menées.
Des stratégies thérapeutiques plus récentes sont en cours d’évaluation. Les mesures générales, comme le contrôle d’un RGO ou l’oxygénothérapie dans les formes avancées, sont indispensables.
Corticothérapie
Du fait des mécanismes inflammatoires évoqués et par analogie avec le traitement de la fibrose pulmonaire idiopathique, la corticothérapie est utilisé dans le traitement de la PIF liée à la ScS. Aucune étude n’est disponible pour évaluer son bénéfice, car elle est utilisée en association avec des immunosuppresseurs. Par ailleurs, le risque de crise rénale sclérodermique associé aux fortes doses de glucocorticoïdes [34, 35], en plus des effets secondaires connus, incitent à n’utiliser que des doses faibles (inférieures à 15mg par jour d’équivalent prednisone).
Immunosuppresseurs
Cyclophosphamide (CYC)
Les premières études suggérant une efficacité du CYC ont été réalisées dans les années 1990 [36-40] et montraient une stabilisation ou une amélioration des EFR. Deux études randomisées contre placebo ont permis de mieux évaluer l’intérêt du CYC dans le traitement de la PIF. La Scleroderma Lung Study [41] a inclus 158 patients atteints de PIF. Les patients traités par CYC (2mg/kg par jour pendant 12 mois) avaient une CVF et une CPT plus élevée que les patients du groupe placebo à 12 mois, mais cette différence était modeste. Aucune différence significative n’a été observée pour la DLCO. En revanche, le suivi à 24 mois ne montrait plus de différence significative entre les 2 groupes [42]. Une autre étude, menée chez 45 patients, randomisés pour recevoir du CYC (6 injections mensuelles) suivi d’un traitement par azathioprine ou un placebo, a montré une tendance à l’amélioration de la CVF dans le groupe traité, sans atteindre une significativité statistique (43).
Le CYC peut être à l’origine d’effets secondaires importants: myélotoxicité, infertilité, infections opportunistes. Ce risque augmente notamment avec la dose cumulée. Pour les limiter, l’utilisation de la voie intraveineuse (IV) a été proposée par plusieurs équipes [39, 40, 43], avec peu d’évènements indésirables graves rapportés.
Les bénéfices apportés par le CYC sont donc modestes. Cependant, une analyse de sous-groupe de la Scleroderma Lung Study montre que le bénéfice du CYC est plus important chez les patients ayant une PIF plus sévère (définie par une CVF < 70% de la valeur théorique). De plus, dans une étude sur 27 patients inclus sur des critères d’aggravation de la PIF dans l’année précédente [44], le traitement par CYC (par voie IV pendant 6 mois, relayé par azathioprine) a permis une amélioration ou stabilisation des EFR qui se maintenait à 2 ans de suivi. Les patients présentant une PIF sévère et/ou évolutive seraient donc les plus susceptibles de bénéficier du traitement par CYC.
Azathioprine (AZA)
Peu de données sont disponibles dans la littérature. Une étude (sur 60 patients) a comparé l’AZA au CYC. Une dégradation des résultats des EFR a été observée dans le groupe AZA [45]. Cette molécule est principalement proposée comme traitement immunosuppresseur d’entretien après le CYC [43, 44].
Mycophénolate Mofetil (MMF)
L’utilisation du mycophénolate mofetyl est récente dans cette indication. Des études retrospectives [46-48] ainsi qu’une étude prospective ouverte sur 5 patients [49] suggèrent une efficacité et une bonne tolérance de ce traitement, mais des études prospectives plus larges sont nécessaires.
Perspectives thérapeutiques
Les essais cliniques randomisés étudiant les anticorps recombinants anti-TGF β1 [50] et un antagoniste des récepteurs de l’endothéline 1 (Bosentan) [51] n’ont pas montré de bénéfices de ces molécules.
Le Rituximab est un anticorps anti-CD20, permettant une déplétion lymphocytaite B. Une étude randomisée sur 14 patients [52] dont 8 ont reçu le rituximab en plus du traitement standard a montré une amélioration de la CVF et de la DLCO par rapport aux patients ayant reçu le traitement standard seul.
L’Imatinib est un inhibiteur de tyrosine kinase, pouvant inhiber la signalisation du TGF β et du CTGF [53]. Dans un essai ouvert sur 30 patients [54] ce traitement a montré une amélioration de l’atteinte cutanée et de la CVF chez les patients traités. Des études randomisées contre placebo seront nécessaires pour mieux évaluer cette stratégie thérapeutique.
Intensification thérapeutique suivie d’autogreffe de cellules souches hématopoïétiques
Depuis la fin des années 1990, l’utilisation de fortes doses d’immunosuppresseurs suivies d’une autogreffe de cellules souches hématopoïétiques (AG-CSH) a été évaluée dans les maladies auto-immunes (55), particulièrement dans la ScS [56-58]. L’interprétation des résultats doit être prudente du fait de l’ absence de groupe contrôle et de l’hétérogénéité des conditionnements(myéloablatif ou non-myéloablatif) et des modalités de recueils des cellules souches. De plus, les patients présentant des atteintes viscérales très sévères ou âgés de plus de 65 ans ne sont pas éligibles en raison de la morbi-mortalité associée à la procédure.
Une étude sur de suivi de 26 patients (suivi médian de 5 ans) montrait une amélioration de l’atteinte cutanée et une stabilisation des atteintes d’organe [59].
Récemment les résultats du premier essai prospectif randomisé, ayant inclus 19 patients, comparant cette technique au traitement immunosuppresseur par CYC ont été publiés [60] et montrent une supériorité de l’intensification suivie d’AG-CSH. Deux autres essais randomisés sont en cours.
Transplantation pulmonaire
Plusieurs études rétrospectives [61-63] ont évalué le bénéfice de la transplantation pulmonaire (TP) chez les patients atteints de ScS. Les patients rapportés dans ces études ont été sélectionnés et des atteintes telles qu’une insuffisance rénale, un RGO sévère non contrôlé par le traitement médical ou des ulcères cutanés non cicatrisés constituaient des contre-indications à la TP.
La survie à 1 et 2 ans des patients transplantés pour une ScS est équivalente à celle des patients transplantés pour d’autres pathologies [62, 63]. Dans l’ étude de Saggar et al. les patients du groupe ScS présentaient significativement plus de rejets aigus mais aucune différence en termes de rejet chronique ou de complications infectieuses comparés aux patients transplantés pour une fibrose pulmonaire idiopathique [62]. Des études de suivi à plus long terme seront nécessaires pour mieux connaitre les bénéfices de la TP.
CONCLUSION
La PIF est une atteinte fréquente de la ScS. Elle est longtemps asymptomatique et la surveillance régulière des patients est indispensable pour la dépister précocement et initier un traitement immunosuppresseur lorsque l’évolution de la maladie est rapide. Les bénéfices des thérapeutiques actuellement utilisées sont limités. L’amélioration des connaissances sur la physiopathologie et le développement de nouvelles stratégies thérapeutiques est indispensable pour améliorer la prise en charge des patients.
CONFLIT D’INTERETS
Aucun.
REFERENCES
1. Jimenez SA, Derk CT. Following the molecular pathways toward an understanding of the pathogenesis of systemic sclerosis. Ann Intern Med 2004;140(1):37-50.
2. Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med. 2009 May 7;360(19):1989-2003.
3. Steen VD, Medsger TA, Jr. Severe organ involvement in systemic sclerosis with diffuse scleroderma. Arthritis Rheum. 2000 Nov;43(11):2437-44.
4. Mayes MD, Lacey JV, Jr., Beebe-Dimmer J, Gillespie BW, Cooper B, Laing TJ, et al. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum. 2003 Aug;48(8):2246-55.
5. Le Guern V, Mahr A, Mouthon L, Jeanneret D, Carzon M, Guillevin L. Prevalence of systemic sclerosis in a French multi-ethnic county. Rheumatology (Oxford). 2004 Sep;43(9):1129-37.
6. Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972-2002. Ann Rheum Dis. 2007 Jul;66(7):940-4.
7. D'Angelo WA, Fries JF, Masi AT, Shulman LE. Pathologic observations in systemic sclerosis (scleroderma). A study of fifty-eight autopsy cases and fifty-eight matched controls. Am J Med. 1969 Mar;46(3):428-40.
8. Ferri C, Valentini G, Cozzi F, Sebastiani M, Michelassi C, La Montagna G, et al. Systemic sclerosis: demographic, clinical, and serologic features and survival in 1,012 Italian patients. Medicine (Baltimore). 2002 Mar;81(2):139-53.
9. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med. 2002 Jan 15;165(2):277-304.
10. Bouros D, Wells AU, Nicholson AG, Colby TV, Polychronopoulos V, Pantelidis P, et al. Histopathologic subsets of fibrosing alveolitis in patients with systemic sclerosis and their relationship to outcome. Am J Respir Crit Care Med. 2002 Jun 15;165(12):1581-6.
11. Kim DS, Yoo B, Lee JS, Kim EK, Lim CM, Lee SD, et al. The major histopathologic pattern of pulmonary fibrosis in scleroderma is nonspecific interstitial pneumonia. Sarcoidosis Vasc Diffuse Lung Dis. 2002 Jun;19(2):1217. 12. Muir TE, Tazelaar HD, Colby TV, Myers JL. Organizing diffuse alveolar damage associated with progressive systemic sclerosis. Mayo Clin Proc. 1997 Jul;72 (7):639-42.
13. Chizzolini C. Update on pathophysiology of scleroderma with special reference to immunoinflammatory events. Ann Med. 2007;39(1):42-53.
14. Chizzolini C, Brembilla NC, Montanari E, Truchetet ME. Fibrosis and immune dysregulation in systemic sclerosis. Autoimmun Rev. 2011 Mar;10(5):276-81.
15. Leask A. Targeting the TGFbeta, endothelin-1 and CCN2 axis to combat fibrosis in scleroderma. Cell Signal. 2008 Aug;20(8):1409-14.
16. Chizzolini C, Rezzonico R, Ribbens C, Burger D, Wollheim FA, Dayer JM. Inhibition of type I collagen production by dermal fibroblasts upon contact with activated T cells: different sensitivity to inhibition between systemic sclerosis and control fibroblasts. Arthritis Rheum. 1998 Nov;41(11):2039-47.
17. Mori Y, Chen SJ, Varga J. Expression and regulation of intracellular SMAD signaling in scleroderma skin fibroblasts. Arthritis Rheum. 2003 Jul;48(7):1964-78.
18. Troshinsky MB, Kane GC, Varga J, Cater JR, Fish JE, Jimenez SA, et al. Pulmonary function and gastroesophageal reflux in systemic sclerosis. Ann Intern Med. 1994 Jul 1;121(1):6-10.
19. Marie I, Dominique S, Levesque H, Ducrotte P, Denis P, Hellot MF, et al. Esophageal involvement and pulmonary manifestations in systemic sclerosis. Arthritis Rheum. 2001 Aug;45(4):346-54.
20. Lock G, Pfeifer M, Straub RH, Zeuner M, Lang B, Scholmerich J, et al. Association of esophageal dysfunction and pulmonary function impairment in systemic sclerosis. Am J Gastroenterol. 1998 Mar;93(3):341-5.
21. Steen VD. Autoantibodies in systemic sclerosis. Semin Arthritis Rheum. 2005 Aug;35(1):35-42.
22. Wells AU, Hansell DM, Rubens MB, King AD, Cramer D, Black CM, et al. Fibrosing alveolitis in systemic sclerosis: indices of lung function in relation to extent of disease on computed tomography. Arthritis Rheum. 1997 Jul;40(7):1229-36.
23. Steen VD, Conte C, Owens GR, Medsger TA, Jr. Severe restrictive lung disease in systemic sclerosis. Arthritis Rheum. 1994 Sep;37(9):1283-9.
24. Plastiras SC, Karadimitrakis SP, Ziakas PD, Vlachoyiannopoulos PG, Moutsopoulos HM, Tzelepis GE. Scleroderma lung: initial forced vital capacity as predictor of pulmonary function decline. Arthritis Rheum. 2006 Aug 15;55(4):598-602.
25. Buch MH, Denton CP, Furst DE, Guillevin L, Rubin LJ, Wells AU, et al. Submaximal exercise testing in the assessment of interstitial lung disease secondary to systemic sclerosis: reproducibility and correlations of the 6-min walk test. Ann Rheum Dis. 2007;66(2):169-73.
26. Garin MC, Highland KB, Silver RM, Strange C. Limitations to the 6-minute walk test in interstitial lung disease and pulmonary hypertension in scleroderma. J Rheumatol. 2009;36(2):330-6.
27. Schurawitzki H, Stiglbauer R, Graninger W, Herold C, Polzleitner D, et al. Interstitial lung disease in progressive systemic sclerosis: high-resolution CT versus radiography. Radiology. 1990;176(3):755-9.
28. Shah RM, Jimenez S, Wechsler R. Significance of ground-glass opacity on HRCT in long-term follow-up of patients with systemic sclerosis. J Thorac Imaging. 2007 May;22(2):120-4.
29. Goh NS, Desai SR, Veeraraghavan S, Hansell DM, Copley SJ, Maher TM, et al. Interstitial lung disease in systemic sclerosis: a simple staging system. Am J Respir Crit Care Med. 2008 Jun 1;177(11):1248-54.
30. Launay D, Remy-Jardin M, Michon-Pasturel U, Mastora I, Hachulla E, Lambert M, et al. High resolution computed tomography in fibrosing alveolitis associated with systemic sclerosis. J Rheumatol. 2006 Sep;33 (9):1789-801.
31. De Santis M, Bosello S, La Torre G, Capuano A, Tolusso B, Pagliari G, et al. Functional, radiological and biological markers of alveolitis and infections of the lower respiratory tract in patients with systemic sclerosis. Respir Res. 2005;6:96.
32. Strange C, Bolster MB, Roth MD, Silver RM, Theodore A, Goldin J, et al. Bronchoalveolar lavage and response to cyclophosphamide in scleroderma interstitial lung disease. Am J Respir Crit Care Med. 2008 Jan 1;177 (1):91-8.
33. Goh NS, Veeraraghavan S, Desai SR, Cramer D, Hansell DM, Denton CP, et al. Bronchoalveolar lavage cellular profiles in patients with systemic sclerosisassociated interstitial lung disease are not predictive of disease progression. Arthritis Rheum. 2007 Jun;56 (6):2005-12.
34. Steen VD, Medsger TA, Jr. Case-control study of corticosteroids and other drugs that either precipitate or protect from the development of scleroderma renal crisis. Arthritis Rheum. 1998 Sep;41(9):1613-9.
35. DeMarco PJ, Weisman MH, Seibold JR, Furst DE, Wong WK, Hurwitz EL, et al. Predictors and outcomes of scleroderma renal crisis: the high-dose versus lowdose D-penicillamine in early diffuse systemic sclerosis trial. Arthritis Rheum. 2002 Nov;46(11):2983-9.
36. Silver RM, Warrick JH, Kinsella MB, Staudt LS, Baumann MH, Strange C. Cyclophosphamide and lowdose prednisone therapy in patients with systemic sclerosis (scleroderma) with interstitial lung disease. J Rheumatol. 1993 May;20(5):838-44.
37. Steen VD, Lanz JK, Jr., Conte C, Owens GR, Medsger TA, Jr. Therapy for severe interstitial lung disease in systemic sclerosis. A retrospective study. Arthritis Rheum. 1994 Sep;37(9):1290-6.
38. Akesson A, Scheja A, Lundin A, Wollheim FA. Improved pulmonary function in systemic sclerosis after treatment with cyclophosphamide. Arthritis Rheum. 1994 May;37(5):729-35.
39. Davas EM, Peppas C, Maragou M, Alvanou E, Hondros D, Dantis PC. Intravenous cyclophosphamide pulse therapy for the treatment of lung disease associated with scleroderma. Clin Rheumatol 1999;18(6):45561.
40. White B, Moore WC, Wigley FM, Xiao HQ, Wise RA. Cyclophosphamide is associated with pulmonary function and survival benefit in patients with scleroderma and alveolitis. Ann Intern Med. 2000;132(12):947-54.
41. Tashkin DP, Elashoff R, Clements PJ, Goldin J, Roth MD, Furst DE, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med. 2006 Jun 22;354(25):2655-66.
42. Tashkin DP, Elashoff R, Clements PJ, Roth MD, Furst DE, Silver RM, et al. Effects of 1-year treatment with cyclophosphamide on outcomes at 2 years in scleroderma lung disease. Am J Respir Crit Care Med. 2007 Nov 15;176(10):1026-34.
43. Hoyles RK, Ellis RW, Wellsbury J, Lees B, Newlands P, Goh NS, et al. A multicenter, prospective, randomized, double-blind, placebo-controlled trial of corticosteroids and intravenous cyclophosphamide followed by oral azathioprine for the treatment of pulmonary fibrosis in scleroderma. Arthritis Rheum. 2006 Dec;54(12):3962-70.
44. Berezne A, Ranque B, Valeyre D, Brauner M, Allanore Y, Launay D, et al. Therapeutic strategy combining intravenous cyclophosphamide followed by oral azathioprine to treat worsening interstitial lung disease associated with systemic sclerosis: a retrospective multicenter open-label study
45. Nadashkevich O, Davis P, Fritzler M, Kovalenko W. A randomized unblinded trial of cyclophosphamide versus azathioprine in the treatment of systemic sclerosis. Clin Rheumatol. 2006 Mar;25(2):205-12.
46. Gerbino AJ, Goss CH, Molitor JA. Effect of mycophenolate mofetil on pulmonary function in sclerodermaassociated interstitial lung disease. Chest 2008;133 (2):455-60.
47. Nihtyanova SI, Brough GM, Black CM, Denton CP. Mycophenolate mofetil in diffuse cutaneous systemic sclerosis--a retrospective analysis. Rheumatology (Oxford). 2007 Mar;46(3):442-5.
48. Zamora AC, Wolters PJ, Collard HR, Connolly MK, Elicker BM, Webb WR, et al. Use of mycophenolate mofetil to treat scleroderma-associated interstitial lung disease. Respir Med. 2008 Jan;102(1):150-5.
49. Liossis SN, Bounas A, Andonopoulos AP. Mycophenolate mofetil as first-line treatment improves clinically evident early scleroderma lung disease. Rheumatology (Oxford). 2006 Aug;45(8):1005-8.
50. Denton CP, Merkel PA, Furst DE, Khanna D, Emery P, Hsu VM, et al. Recombinant human anti-transforming growth factor beta1 antibody therapy in systemic sclerosis: a multicenter, randomized, placebo-controlled phase I/II trial of CAT-192. Arthritis Rheum. 2007 Jan;56(1):323-33.
51. Seibold JR, Denton CP, Furst DE, Guillevin L, Rubin LJ, Wells A, et al. Randomized, prospective, placebocontrolled trial of bosentan in interstitial lung disease secondary to systemic sclerosis. Arthritis Rheum. 2010 Jul;62(7):2101-8.
52. Daoussis D, Liossis SN, Tsamandas AC, Kalogeropoulou C, Kazantzi A, Sirinian C, et al. Experience with rituximab in scleroderma: results from a 1-year, proofof-principle study. Rheumatology (Oxford). 2010 Feb;49(2):271-80.
53. Distler JH, Distler O. Tyrosine kinase inhibitors for the treatment of fibrotic diseases such as systemic sclerosis: towards molecular targeted therapies. Ann Rheum Dis. 2010 Jan;69 Suppl 1:i48-51.
54. Spiera RF, Gordon JK, Mersten JN, Magro CM, Mehta M, Wildman HF, et al. Imatinib mesylate (Gleevec) in the treatment of diffuse cutaneous systemic sclerosis: results of a 1-year, phase IIa, single-arm, open-label clinical trial. Ann Rheum Dis. 2011 Jun;70(6):1003-9.
55. Tyndall A, Fassas A, Passweg J, Ruiz de Elvira C, Attal M, Brooks P, et al. Autologous haematopoietic stem cell transplants for autoimmune disease--feasibility and transplant-related mortality. Autoimmune Disease and Lymphoma Working Parties of the European Group for Blood and Marrow Transplantation, the European League Against Rheumatism and the International Stem Cell Project for Autoimmune Disease. Bone Marrow Transplant. 1999 Oct;24(7):729-34.
56. Farge D, Passweg J, van Laar JM, Marjanovic Z, Besenthal C, Finke J, et al. Autologous stem cell transplantation in the treatment of systemic sclerosis: report from the EBMT/EULAR Registry. Ann Rheum Dis. 2004 Aug;63(8):974-81.
57. McSweeney PA, Nash RA, Sullivan KM, Storek J, Crofford LJ, Dansey R, et al. High-dose immunosuppressive therapy for severe systemic sclerosis: initial outcomes. Blood. 2002 Sep 1;100(5):1602-10.
58. Binks M, Passweg JR, Furst D, McSweeney P, Sullivan K, Besenthal C, et al. Phase I/II trial of autologous stem cell transplantation in systemic sclerosis: procedure related mortality and impact on skin disease. Ann Rheum Dis. 2001 Jun;60(6):577-84.
59. Vonk MC, Marjanovic Z, van den Hoogen FH, Zohar S, Schattenberg AV, Fibbe WE, et al. Long-term follow-up results after autologous haematopoietic stem cell transplantation for severe systemic sclerosis. Ann Rheum Dis. 2008 Jan;67(1):98-104.
60. Burt RK, Shah SJ, Dill K, Grant T, Gheorghiade M, Schroeder J, et al. Autologous non-myeloablative haemopoietic stem-cell transplantation compared with pulse cyclophosphamide once per month for systemic sclerosis (ASSIST): an open-label, randomised phase 2 trial. Lancet. 2011 Aug 6;378(9790):498-506.
61. Massad MG, Powell CR, Kpodonu J, Tshibaka C, Hanhan Z, Snow NJ, et al. Outcomes of lung transplantation in patients with scleroderma. World J Surg. 2005 Nov;29(11):1510-5.
62. Saggar R, Khanna D, Furst DE, Belperio JA, Park GS, Weigt SS, et al. Systemic sclerosis and bilateral lung transplantation: a single centre experience. Eur Respir J. 2010 Oct;36(4):893-900.
63. Schachna L, Medsger TA, Jr., Dauber JH, Wigley FM, Braunstein NA, White B, et al. Lung transplantation in scleroderma compared with idiopathic pulmonary fibrosis and idiopathic pulmonary arterial hypertension. Arthritis Rheum. 2006 Dec;54(12):3954-61.

FIGURES
REFERENCES
1. Jimenez SA, Derk CT. Following the molecular pathways toward an understanding of the pathogenesis of systemic sclerosis. Ann Intern Med 2004;140(1):37-50.
2. Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med. 2009 May 7;360(19):1989-2003.
3. Steen VD, Medsger TA, Jr. Severe organ involvement in systemic sclerosis with diffuse scleroderma. Arthritis Rheum. 2000 Nov;43(11):2437-44.
4. Mayes MD, Lacey JV, Jr., Beebe-Dimmer J, Gillespie BW, Cooper B, Laing TJ, et al. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum. 2003 Aug;48(8):2246-55.
5. Le Guern V, Mahr A, Mouthon L, Jeanneret D, Carzon M, Guillevin L. Prevalence of systemic sclerosis in a French multi-ethnic county. Rheumatology (Oxford). 2004 Sep;43(9):1129-37.
6. Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972-2002. Ann Rheum Dis. 2007 Jul;66(7):940-4.
7. D'Angelo WA, Fries JF, Masi AT, Shulman LE. Pathologic observations in systemic sclerosis (scleroderma). A study of fifty-eight autopsy cases and fifty-eight matched controls. Am J Med. 1969 Mar;46(3):428-40.
8. Ferri C, Valentini G, Cozzi F, Sebastiani M, Michelassi C, La Montagna G, et al. Systemic sclerosis: demographic, clinical, and serologic features and survival in 1,012 Italian patients. Medicine (Baltimore). 2002 Mar;81(2):139-53.
9. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med. 2002 Jan 15;165(2):277-304.
10. Bouros D, Wells AU, Nicholson AG, Colby TV, Polychronopoulos V, Pantelidis P, et al. Histopathologic subsets of fibrosing alveolitis in patients with systemic sclerosis and their relationship to outcome. Am J Respir Crit Care Med. 2002 Jun 15;165(12):1581-6.
11. Kim DS, Yoo B, Lee JS, Kim EK, Lim CM, Lee SD, et al. The major histopathologic pattern of pulmonary fibrosis in scleroderma is nonspecific interstitial pneumonia. Sarcoidosis Vasc Diffuse Lung Dis. 2002 Jun;19(2):1217. 12. Muir TE, Tazelaar HD, Colby TV, Myers JL. Organizing diffuse alveolar damage associated with progressive systemic sclerosis. Mayo Clin Proc. 1997 Jul;72 (7):639-42.
13. Chizzolini C. Update on pathophysiology of scleroderma with special reference to immunoinflammatory events. Ann Med. 2007;39(1):42-53.
14. Chizzolini C, Brembilla NC, Montanari E, Truchetet ME. Fibrosis and immune dysregulation in systemic sclerosis. Autoimmun Rev. 2011 Mar;10(5):276-81.
15. Leask A. Targeting the TGFbeta, endothelin-1 and CCN2 axis to combat fibrosis in scleroderma. Cell Signal. 2008 Aug;20(8):1409-14.
16. Chizzolini C, Rezzonico R, Ribbens C, Burger D, Wollheim FA, Dayer JM. Inhibition of type I collagen production by dermal fibroblasts upon contact with activated T cells: different sensitivity to inhibition between systemic sclerosis and control fibroblasts. Arthritis Rheum. 1998 Nov;41(11):2039-47.
17. Mori Y, Chen SJ, Varga J. Expression and regulation of intracellular SMAD signaling in scleroderma skin fibroblasts. Arthritis Rheum. 2003 Jul;48(7):1964-78.
18. Troshinsky MB, Kane GC, Varga J, Cater JR, Fish JE, Jimenez SA, et al. Pulmonary function and gastroesophageal reflux in systemic sclerosis. Ann Intern Med. 1994 Jul 1;121(1):6-10.
19. Marie I, Dominique S, Levesque H, Ducrotte P, Denis P, Hellot MF, et al. Esophageal involvement and pulmonary manifestations in systemic sclerosis. Arthritis Rheum. 2001 Aug;45(4):346-54.
20. Lock G, Pfeifer M, Straub RH, Zeuner M, Lang B, Scholmerich J, et al. Association of esophageal dysfunction and pulmonary function impairment in systemic sclerosis. Am J Gastroenterol. 1998 Mar;93(3):341-5.
21. Steen VD. Autoantibodies in systemic sclerosis. Semin Arthritis Rheum. 2005 Aug;35(1):35-42.
22. Wells AU, Hansell DM, Rubens MB, King AD, Cramer D, Black CM, et al. Fibrosing alveolitis in systemic sclerosis: indices of lung function in relation to extent of disease on computed tomography. Arthritis Rheum. 1997 Jul;40(7):1229-36.
23. Steen VD, Conte C, Owens GR, Medsger TA, Jr. Severe restrictive lung disease in systemic sclerosis. Arthritis Rheum. 1994 Sep;37(9):1283-9.
24. Plastiras SC, Karadimitrakis SP, Ziakas PD, Vlachoyiannopoulos PG, Moutsopoulos HM, Tzelepis GE. Scleroderma lung: initial forced vital capacity as predictor of pulmonary function decline. Arthritis Rheum. 2006 Aug 15;55(4):598-602.
25. Buch MH, Denton CP, Furst DE, Guillevin L, Rubin LJ, Wells AU, et al. Submaximal exercise testing in the assessment of interstitial lung disease secondary to systemic sclerosis: reproducibility and correlations of the 6-min walk test. Ann Rheum Dis. 2007;66(2):169-73.
26. Garin MC, Highland KB, Silver RM, Strange C. Limitations to the 6-minute walk test in interstitial lung disease and pulmonary hypertension in scleroderma. J Rheumatol. 2009;36(2):330-6.
27. Schurawitzki H, Stiglbauer R, Graninger W, Herold C, Polzleitner D, et al. Interstitial lung disease in progressive systemic sclerosis: high-resolution CT versus radiography. Radiology. 1990;176(3):755-9.
28. Shah RM, Jimenez S, Wechsler R. Significance of ground-glass opacity on HRCT in long-term follow-up of patients with systemic sclerosis. J Thorac Imaging. 2007 May;22(2):120-4.
29. Goh NS, Desai SR, Veeraraghavan S, Hansell DM, Copley SJ, Maher TM, et al. Interstitial lung disease in systemic sclerosis: a simple staging system. Am J Respir Crit Care Med. 2008 Jun 1;177(11):1248-54.
30. Launay D, Remy-Jardin M, Michon-Pasturel U, Mastora I, Hachulla E, Lambert M, et al. High resolution computed tomography in fibrosing alveolitis associated with systemic sclerosis. J Rheumatol. 2006 Sep;33 (9):1789-801.
31. De Santis M, Bosello S, La Torre G, Capuano A, Tolusso B, Pagliari G, et al. Functional, radiological and biological markers of alveolitis and infections of the lower respiratory tract in patients with systemic sclerosis. Respir Res. 2005;6:96.
32. Strange C, Bolster MB, Roth MD, Silver RM, Theodore A, Goldin J, et al. Bronchoalveolar lavage and response to cyclophosphamide in scleroderma interstitial lung disease. Am J Respir Crit Care Med. 2008 Jan 1;177 (1):91-8.
33. Goh NS, Veeraraghavan S, Desai SR, Cramer D, Hansell DM, Denton CP, et al. Bronchoalveolar lavage cellular profiles in patients with systemic sclerosisassociated interstitial lung disease are not predictive of disease progression. Arthritis Rheum. 2007 Jun;56 (6):2005-12.
34. Steen VD, Medsger TA, Jr. Case-control study of corticosteroids and other drugs that either precipitate or protect from the development of scleroderma renal crisis. Arthritis Rheum. 1998 Sep;41(9):1613-9.
35. DeMarco PJ, Weisman MH, Seibold JR, Furst DE, Wong WK, Hurwitz EL, et al. Predictors and outcomes of scleroderma renal crisis: the high-dose versus lowdose D-penicillamine in early diffuse systemic sclerosis trial. Arthritis Rheum. 2002 Nov;46(11):2983-9.
36. Silver RM, Warrick JH, Kinsella MB, Staudt LS, Baumann MH, Strange C. Cyclophosphamide and lowdose prednisone therapy in patients with systemic sclerosis (scleroderma) with interstitial lung disease. J Rheumatol. 1993 May;20(5):838-44.
37. Steen VD, Lanz JK, Jr., Conte C, Owens GR, Medsger TA, Jr. Therapy for severe interstitial lung disease in systemic sclerosis. A retrospective study. Arthritis Rheum. 1994 Sep;37(9):1290-6.
38. Akesson A, Scheja A, Lundin A, Wollheim FA. Improved pulmonary function in systemic sclerosis after treatment with cyclophosphamide. Arthritis Rheum. 1994 May;37(5):729-35.
39. Davas EM, Peppas C, Maragou M, Alvanou E, Hondros D, Dantis PC. Intravenous cyclophosphamide pulse therapy for the treatment of lung disease associated with scleroderma. Clin Rheumatol 1999;18(6):45561.
40. White B, Moore WC, Wigley FM, Xiao HQ, Wise RA. Cyclophosphamide is associated with pulmonary function and survival benefit in patients with scleroderma and alveolitis. Ann Intern Med. 2000;132(12):947-54.
41. Tashkin DP, Elashoff R, Clements PJ, Goldin J, Roth MD, Furst DE, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med. 2006 Jun 22;354(25):2655-66.
42. Tashkin DP, Elashoff R, Clements PJ, Roth MD, Furst DE, Silver RM, et al. Effects of 1-year treatment with cyclophosphamide on outcomes at 2 years in scleroderma lung disease. Am J Respir Crit Care Med. 2007 Nov 15;176(10):1026-34.
43. Hoyles RK, Ellis RW, Wellsbury J, Lees B, Newlands P, Goh NS, et al. A multicenter, prospective, randomized, double-blind, placebo-controlled trial of corticosteroids and intravenous cyclophosphamide followed by oral azathioprine for the treatment of pulmonary fibrosis in scleroderma. Arthritis Rheum. 2006 Dec;54(12):3962-70.
44. Berezne A, Ranque B, Valeyre D, Brauner M, Allanore Y, Launay D, et al. Therapeutic strategy combining intravenous cyclophosphamide followed by oral azathioprine to treat worsening interstitial lung disease associated with systemic sclerosis: a retrospective multicenter open-label study
45. Nadashkevich O, Davis P, Fritzler M, Kovalenko W. A randomized unblinded trial of cyclophosphamide versus azathioprine in the treatment of systemic sclerosis. Clin Rheumatol. 2006 Mar;25(2):205-12.
46. Gerbino AJ, Goss CH, Molitor JA. Effect of mycophenolate mofetil on pulmonary function in sclerodermaassociated interstitial lung disease. Chest 2008;133 (2):455-60.
47. Nihtyanova SI, Brough GM, Black CM, Denton CP. Mycophenolate mofetil in diffuse cutaneous systemic sclerosis--a retrospective analysis. Rheumatology (Oxford). 2007 Mar;46(3):442-5.
48. Zamora AC, Wolters PJ, Collard HR, Connolly MK, Elicker BM, Webb WR, et al. Use of mycophenolate mofetil to treat scleroderma-associated interstitial lung disease. Respir Med. 2008 Jan;102(1):150-5.
49. Liossis SN, Bounas A, Andonopoulos AP. Mycophenolate mofetil as first-line treatment improves clinically evident early scleroderma lung disease. Rheumatology (Oxford). 2006 Aug;45(8):1005-8.
50. Denton CP, Merkel PA, Furst DE, Khanna D, Emery P, Hsu VM, et al. Recombinant human anti-transforming growth factor beta1 antibody therapy in systemic sclerosis: a multicenter, randomized, placebo-controlled phase I/II trial of CAT-192. Arthritis Rheum. 2007 Jan;56(1):323-33.
51. Seibold JR, Denton CP, Furst DE, Guillevin L, Rubin LJ, Wells A, et al. Randomized, prospective, placebocontrolled trial of bosentan in interstitial lung disease secondary to systemic sclerosis. Arthritis Rheum. 2010 Jul;62(7):2101-8.
52. Daoussis D, Liossis SN, Tsamandas AC, Kalogeropoulou C, Kazantzi A, Sirinian C, et al. Experience with rituximab in scleroderma: results from a 1-year, proofof-principle study. Rheumatology (Oxford). 2010 Feb;49(2):271-80.
53. Distler JH, Distler O. Tyrosine kinase inhibitors for the treatment of fibrotic diseases such as systemic sclerosis: towards molecular targeted therapies. Ann Rheum Dis. 2010 Jan;69 Suppl 1:i48-51.
54. Spiera RF, Gordon JK, Mersten JN, Magro CM, Mehta M, Wildman HF, et al. Imatinib mesylate (Gleevec) in the treatment of diffuse cutaneous systemic sclerosis: results of a 1-year, phase IIa, single-arm, open-label clinical trial. Ann Rheum Dis. 2011 Jun;70(6):1003-9.
55. Tyndall A, Fassas A, Passweg J, Ruiz de Elvira C, Attal M, Brooks P, et al. Autologous haematopoietic stem cell transplants for autoimmune disease--feasibility and transplant-related mortality. Autoimmune Disease and Lymphoma Working Parties of the European Group for Blood and Marrow Transplantation, the European League Against Rheumatism and the International Stem Cell Project for Autoimmune Disease. Bone Marrow Transplant. 1999 Oct;24(7):729-34.
56. Farge D, Passweg J, van Laar JM, Marjanovic Z, Besenthal C, Finke J, et al. Autologous stem cell transplantation in the treatment of systemic sclerosis: report from the EBMT/EULAR Registry. Ann Rheum Dis. 2004 Aug;63(8):974-81.
57. McSweeney PA, Nash RA, Sullivan KM, Storek J, Crofford LJ, Dansey R, et al. High-dose immunosuppressive therapy for severe systemic sclerosis: initial outcomes. Blood. 2002 Sep 1;100(5):1602-10.
58. Binks M, Passweg JR, Furst D, McSweeney P, Sullivan K, Besenthal C, et al. Phase I/II trial of autologous stem cell transplantation in systemic sclerosis: procedure related mortality and impact on skin disease. Ann Rheum Dis. 2001 Jun;60(6):577-84.
59. Vonk MC, Marjanovic Z, van den Hoogen FH, Zohar S, Schattenberg AV, Fibbe WE, et al. Long-term follow-up results after autologous haematopoietic stem cell transplantation for severe systemic sclerosis. Ann Rheum Dis. 2008 Jan;67(1):98-104.
60. Burt RK, Shah SJ, Dill K, Grant T, Gheorghiade M, Schroeder J, et al. Autologous non-myeloablative haemopoietic stem-cell transplantation compared with pulse cyclophosphamide once per month for systemic sclerosis (ASSIST): an open-label, randomised phase 2 trial. Lancet. 2011 Aug 6;378(9790):498-506.
61. Massad MG, Powell CR, Kpodonu J, Tshibaka C, Hanhan Z, Snow NJ, et al. Outcomes of lung transplantation in patients with scleroderma. World J Surg. 2005 Nov;29(11):1510-5.
62. Saggar R, Khanna D, Furst DE, Belperio JA, Park GS, Weigt SS, et al. Systemic sclerosis and bilateral lung transplantation: a single centre experience. Eur Respir J. 2010 Oct;36(4):893-900.
63. Schachna L, Medsger TA, Jr., Dauber JH, Wigley FM, Braunstein NA, White B, et al. Lung transplantation in scleroderma compared with idiopathic pulmonary fibrosis and idiopathic pulmonary arterial hypertension. Arthritis Rheum. 2006 Dec;54(12):3954-61.
ARTICLE INFO
DOI: 10.12699/jfvp.2.5.2011.13
Conflict of Interest
Non
Date of manuscript receiving
27/3/2011
Date of publication after correction
16/9/2011
Article citation
Rivière S. Interstitial lung disease due to systemic sclerosis. J Func Vent Pulm 2011;02(05):13-20.



Copyright: jfvpulm.com