
 English
English
 French
French
 Pubmed
Pubmed Google Scholar
Google Scholar Cross Ref
Cross Ref Visitor 725689
Visitor 725689Dysfonctionnement endothélial dans la BPCO: Rôle du monoxyde d’azote et d’endothéline-1
Endothelial dysfunction in COPD: Role of nitric oxide and endothelin-1
S. Duong-Quy, N.N. Le-Dong, T. Hua-Huy, A.T. Dinh-Xuan
Service de Physiologie-Explorations Fonctionnelles Cardio-Respiratoires. Hôpital Cochin - Paris
Laboratoire Biologie - Physiologie des Maladies Respiraoires. UPRES - EA 2511. Université Paris Descartes
Corresponding author
Dr Sy DUONG-QUY
Service de Physiologie - EFR. Pôle Cardio-Resiratoires. Hôpital Cochin
E-mail: sy.duong-quy@cch.aphp.fr
ABSTRACT
Cigarette smoke contains many toxic components of which most diffuse through the alveolar capillary barrier and pass into pulmonary circulation. However, the direct and indirect effects of cigarette smoke on pulmonary endothelial dysfunction are still poorly understood. It is also reported that endothelium-dependent vasorelaxation was significantly reduced in patients with COPD.
Recent studies showed that the structural changes of pulmonary arteries of patients with COPD have been implicated in pulmonary endothelial dysfunction. These data suggest the role of cigarette smoke in the structural and functional changes of endothelium.
In general, the endothelium acts not only as an interface between the blood flow and vascular smooth muscle cells, it also plays a fundamental role in the regulation of vascular tone via vasoactive agents. These substances, synthesized by endothelial cells, react in a paracrin manner on the underlying smooth muscle cells to induce the relaxation via nitric oxide (NO), or conversely, the contraction via endothelin-1 (ET-1), a potent vasoconstrictor and mitogenic agent.
In patient with COPD post-smoking, pulmonary endothelial dysfunction might be related to the decrease of NOS-3 (endothelial nitric oxide synthase) activity in the production of NO or the increase of ET-1 system activity.
KEYWORDS: COPD, endothelial dysfunction, cigarette smoke, NO, ET-1
RÉSUMÉ
La fumée de tabac contient plusieurs composants toxiques dont une majeure partie diffusent à travers la barrière capillaire alvéolaire et passent dans la circulation pulmonaire. Cependant, les effets directs et indirects de la fumée du tabac sur le dysfonctionnement endothéliale pulmonaire est encore peu étudiés. Il a été rapporté que la vasorelaxation dépendante de l’endothélium est significativement diminuée chez les patients atteints de BPCO.
Des études récentes ont montré que des altérations de la structure des artères pulmonaires, retrouvées chez des patients avec BPCO, ont été impliquées dans le dysfonctionnement endothélial. Ces données suggèrent le rôle de la fumée de cigarette dans la modification structurale et fonctionnelle de l’endothélium.
En général, l’endothélium remplit non seulement le rôle d’interface entre le flux sanguin et les cellules musculaires lisses vasculaires, il joue également un rôle fondamental dans la régulation du tonus vasculaire par l'intermédiaire d’agents vasoactifs. Ces substances, synthétisées par les cellules endothéliales, agissent de façon paracrine sur les cellules musculaires lisses sous jacente en induisant une relaxation via le monoxyde d’azote (NO) ou, au contraire une contraction via l’endothéline-1 (ET-1), un puissant vasoconstricteur et un agent mitogénique.
Donc, chez les BPCO post-tabagiques, le dysfonctionnement endothélial pulmonaire pourrait être lié à la diminution de l’activité de la NOS-3 (endothelial nitric oxide synthase) dans la production du NO ou à l’augmentation de l’activité du système d’ET-1.
MOTS CLES: BPCO, dysfonctionnement endothélial, fumée de cigarette, NO, ET-1
INTRODUCTION
La prévalence de la broncho-pneumopathie chronique obstructive (BPCO) est en augmentation constante depuis 20 ans partout dans le monde. Actuellement, on estime à plus de 44 millions de sujets porteurs de BPCO, soit de 4 à 10% de la population adulte mondiale. La mortalité liée à la BPCO devrait doubler en 30 ans (de 1990 à 2020). On estime que la BPCO sera la 3ème cause de mortalité (après les cardiopathies ischémiques et les maladies vasculaires cérébrales) en 2020. Ceci sera en grande partie dû à l’augmentation du tabagisme notamment chez les femmes [1, 2].
DYSFONCTIONNEMENT ENDOTHELIAL DANS LA BPCO POST-TABAGIQUE
Rôle du tabagisme dans le dysfonctionnement endothélial
Bien que l’atteinte des voies aériennes causée par le tabac soit bien connue, les effets directs et indirects de la fumée du tabac sur la fonction endothéliale pulmonaire ont été relativement peu étudiés. En effet, les fumeurs actifs atteints de BPCO ont un trouble de la relaxation artérielle pulmonaire lié au dysfonctionnement des cellules endothéliales [3].
La fumée de tabac contient plus de 4 000 composants dont une majeure partie diffusent à travers la barrière capillaire alvéolaire et passent immédiatement dans la circulation pulmonaire [4]. Le dysfonctionnement endothélial des vaisseaux pulmonaires causé par le tabac reste en partie inexpliqué. Cependant il est admis que l’absorption des composants du tabac entraîne une altération de la fonction des cellules endothéliales vasculaires pulmonaires [5]. Il a été rapporté que la vasorelaxation dépendante de l’ endothélium est diminuée chez les fumeurs ainsi que chez les patients atteints de BPCO [6]. De plus il est établi que les fumeurs présentent une diminution de la concentration de NO dans l’air expiré, suggérant une diminution de la production de NO lié au tabagisme de ces sujets [3].
Lésions vasculaires pulmonaires dans la BPCO
L’étude histologique des vaisseaux pulmonaires des patients atteints de BPCO, montre un épaississement de l’intima, en rapport avec une prolifération des cellules musculaires lisses de la média, et à des dépôts de fibres de collagène et élastiques. L’activation de l’endothélium par des médiateurs inflammatoires se manifeste notamment avec l’augmentation de l’expression de molécules d’adhésion.
L’hypoxie chronique est à la fois la conséquence de l’obstruction bronchique et de l’hypoventilation alvéolaire caractérisant les BPCO à un stade avancé et la cause de la constriction et le remodelage des vaisseaux pulmonaires [7]. L’autopsie des malades atteints de BPCO sévère et fatale a montré l’existence de lésions au niveau des muscles lisses des artères et des artérioles pré-capillaires de la circulation pulmonaire [8, 9]. Les anomalies structurales des vaisseaux pulmonaires de ces sujets sont caractérisées par une prolifération excessive des cellules musculaires lisses et par un dépôt d’élastine et de fibres de collagène responsables du rétrécissement de la lumière des artères [10].
L’hypoxémie chronique, impliquée dans la genèse de ces lésions, n’est pas seule responsable et un épaississement de l’intima peut se voir chez des patients fumeurs non hypoxémiques atteints de BPCO légère. D’autres facteurs sont impliqués, comme la réaction inflammatoire, mise en évidence par une augmentation des lymphocytes CD8+ retrouvés dans l’adventice des vaisseaux pulmonaires de patients fumeurs, BPCO ou non [11]. Il semble donc que la fumée de cigarette puisse être directement responsable de lésions de l’endothélium vasculaire pulmonaire, ces lésions pouvantêtre majorées par l’hypoxémie et l’inflammation chronique.
De plus, l’hypoxie chronique est connue pour être à l’origine d’une hypertension artérielle pulmonaire (HTAP), même si celle-ci reste souvent modérée, chez la plupart des patients porteurs d’une BPCO avec hypoxémie [8, 12]. Des études récentes ont montré que des altérations de la structure des artères pulmonaires peuvent également être retrouvées chez des patients avec BPCO post-tabagique sans hypoxémie, voire chez certains fumeurs dont la fonction respiratoire est encore parfaitement normale [13, 14]. Ces données suggèrent un rôle de la fumée de cigarette dans la modification tant structurale que fonctionnelle des vaisseaux pulmonaires, portant notamment sur la structure et la fonction de la cellule endothéliale.
ROLE DU MONOXYDE D’AZOTE (NO) ET ENDOTHELINE-1 (ET-1) DANS LE DYSFONCTIONNEMENTENDOTHELIAL CHEZ LES BPCO POST-TABAGIQUES
L’endothélium remplit non seulement le rôle d’interface entre le flux sanguin et la cellule musculaire lisse vasculaire, il joue également un rôle fondamental dans la régulation du tonus vasculaire par l'intermédiaire d’agents vasoactifs. Ces substances synthétisées par la cellule endothéliale agissent de façon paracrine sur la cellule musculaire lisse sous jacente en induisant une relaxation, comme le monoxyde d’azote (NO) ou, au contraire une contraction, comme l’endothéline-1. Dans la cellule musculaire lisse vasculaire, le tonus (autrement dit l’état de contractilité) est principalement lié à la concentration d’un second messager nucléotidique, le guanosine monophosphate cyclique (GMPc). Celui-ci, comme son congénère mieux connu et plus célèbre, l’adénosine monophosphate cyclique (AMPc), est synthétisé sous l’action d’enzymes catalytiques, les guanylate cyclase pour les premiers, les adénylates cyclase pour les seconds. Il existe deux familles de guanylate cyclase, les guanylate cyclase membranaires, récepteurs des peptides natriurétiques et les guanylate cyclase solubles (GCs), principal enzyme intracytosolique responsable de la production du GMPc dans la cellule musculaire lisse des vaisseaux [15].
Rôle du monoxyde d’azote (NO)
Structure moléculaire du NO
Découvert en 1980, et identifié initialement comme facteur relaxant dérivé de l’endothélium (EDRF, endothelium-derived relaxing factor) par Furchgott et Zawadzki [16], le NO joue un rôle physiologique important dans de nombreux organes. Son implication dans diverses maladies humaines a été également démontrée.
Le NO est avant tout un gaz, qui possède à la fois des propriétés chimiques similaires à celles des radicaux libres (oxygénés ou azotés) et des caractéristiques d’un médiateur cellulaire assurant la communication entre deux cellules avoisinantes (communication paracrine intrercellulaire) ou à l’ intérieur d’une même cellule (communication intracellulaire).
Au niveau des appareils circulatoire et respiratoire, le NO joue un rôle essentiel dans la régulation des fonctions physiologiques de ces organes allant de la modulation des tonus vasculaires et bronchiques jusqu’à servir de support moléculaire à l’immunité non spécifique et la transmission synaptique du système non-adrénergique non-cholinergique.
Le NO est un gaz incolore et inodore. Il est peu stable car se combinant facilement avec des radicaux libres, notamment l’anion superoxyde (O2-) pour former l’anion peroxynitrite (ONOO-). L'association d'un atome d'azote avec un atome d'oxygène laisse un électron non apparié, cet électron isolé confère au NO les propriétés d’un radical très réactif, qui franchit les membranes biologiques en réagissant rapidement avec d'autres substances. La demi-vie du NO est de 5 à 6 millisecondes. En raison de sa forte capacité de diffusion, le NO peut non seulement traverser tous les compartiments cellulaires, mais il peut aussi créer un champ d'action autour des cellules qui le produisent.
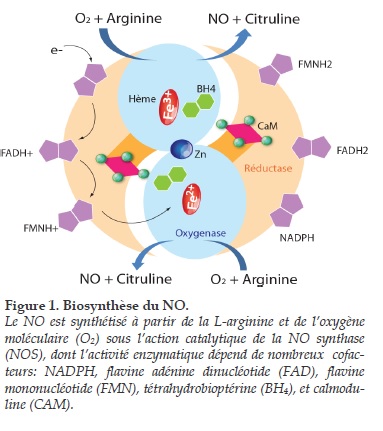
Le NO est synthétisé à partir de l'un des deux atomes d'azote terminal chimiquement équivalents du groupement guanidine de la L-arginine, d’une part, et de l'oxygène moléculaire (O2), d’autre part [17] (Figure 1).
L'autre produit de synthèse, formé de manière stœchiométrique avec le NO, est la L-citrulline, qui dérive de la L-arginine, d'abord hydroxylée en NG-hydroxy-L-arginine. La réaction de biosynthèse du NO et de la L-citrulline à partir de la L-arginine et de l'O2 est sous la dépendance d'une famille d'enzymes, les NOS, dont il existe au moins trois isoformes. Ces trois isoformes - codées par trois gènes distincts localisés sur les chromosomes 7, 12 et 17 – diffèrent entre elles par leurs localisations cellulaires, leurs fonctions et leurs caractéristiques biochimiques.
Les isoformes présentes dans les cellules endothéliales (NOSe ou NOS-3), d'une part, et les cellules nerveuses (NOSn ou NOS-1), d'autre part, appartiennent à la famille des NOS constitutives, c'est-à-dire celles dont l'expression - normalement présente à l'état physiologique – permet la synthèse du NO respectivement en tant que médiateur paracrine de la relaxation du muscle lisse vasculaire et en tant que neurotransmetteur. A l'inverse, l'isoforme macrophagique appartient à la famille des NOS inductibles (NOSi), c'est-à-dire celles dont l'expression normalement absente à l'état physiologique ne se manifeste que dans des états pathologiques. La NOS-3 est activée principalement par les forces de cisaillement ou par la stimulation des récepteurs membranaires par des agonistes entraînant une augmentation de la concentration de calcium à l’intérieur de la cellule endothéliale.
Bien que normalement présente dans la cellule (endothéliale ou neuronale), l'isoforme constitutive de la NOS n'est pas active en l'absence d'une augmentation transitoire du calcium intracellulaire et de l'activation de la calmoduline qui en résulte.
L'activité de la NOS constitutive se traduit par la production d'une faible quantité de NO pendant une période brève. A l'opposé, l'induction du gène codant la NOSi donne lieu à la synthèse de novo de cette protéine par activation transcriptionnelle. La NOSi, une fois produite par la traduction de l’ARN messager en protéine, devient continuellement active du fait de sa liaison quasi irréversible à la calmoduline. Ceci explique la relative indépendance de cette isoforme par rapport au calcium intracellulaire.
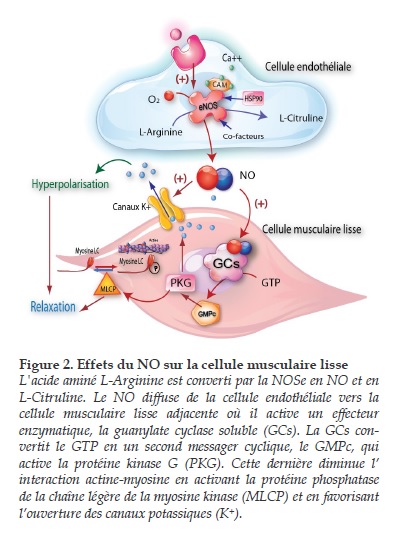
Une fois synthétisé, le NO diffuse rapidement vers l'extérieur de la cellule qui le synthétise et agit sur les cellules avoisinantes selon un mode paracrine. Dans la cellule du muscle lisse vasculaire sous jacente à la cellule endothéliale, le NO agit en stimulant la guanylate cyclase soluble qui synthétise à son tour un second messager nucléotidique le guanosine monophosphate cyclique (GMPc). Le GMPc active la protéine kinase G (PKG), dont les phosphorylations sur diverses molécules cibles contrôlant l’interaction actine myosine aboutissent à la vasodilatation (Figure 2).
Rôle du NO dans la physiopathologie de la BPCO
Les cellules endothéliales vasculaires pulmonaires humaines, où la NOS-3 est fortement exprimée, sont une source importante de NO [3]. L’inhibition de l’activité de la NOS-3 dans les cellules endothéliales peut expliquer la diminution de la vasodilatation dépendante de l'endothélium observée chez les fumeurs avec BPCO ; ainsi que la diminution de la concentration de NO dans l’air expiré de ces fumeurs.
Su et al. ont montré que l’activité de la NOS-3 est diminuée dans les cellules endothéliales porcines exposées à la fumée de tabac [18]. La diminution de la concentration de NO exhalé mesurée chez les fumeurs peut s’expliquer par plusieurs mécanismes: il peut s’agir soit d’une inhibition de l’activité de la NOS liée à la forte quantité de NO présente dans la fumée de cigarette (le NO est connu pour sa capacité d’inhiber sa propre production), soit d’une inactivation du NO par les composés oxydants contenus dans la fumée de cigarette.
Le rôle du NO a été incriminé dans la physiopathologie de la BPCO, notamment par le pouvoir pro-oxydatif du NO et des autres radicaux libres contenus dans la fumée de tabac. L’expression du TNFα, cytokine pro-inflammatoire connue pour augmenter la transcription du gène codant NOS-2 (ou NOSi) dans l’épithélium de voies respiratoires, est augmentée chez les patients BPCO suggérant le rôle de la forme inductible de la NOS dans la progression de la maladie [19, 20].
La diminution de la production de NO suite à l’inhibition de la NOS-3 endothéliale peut être, au moins en partie, responsable de l’augmentation du risque de maladie vasculaire pulmonaire chez certains fumeurs, et de la survenue de l’HTAP associée à la BPCO due au tabac.
Les endothelines
Biosynthèse des endothélines
La famille des endothélines comprend trois peptides: endothéline-1 (ET-1), endothéline-2 (ET-2) et endothéline-3 (ET-3). Ces 3 peptides ont tous 21 acides aminés, dont les séquences ne diffèrent entre elles que sur quelques résidus. Les principales particularités entre ces trois isoformes tiennent de leur profil d’expression tissulaire et de leur affinité pour les deux récepteurs membranaires de l’endothéline: le récepteur ET-A et le récepteur ET-B.
Les endothélines sont synthétisées principalement par les cellules endothéliales mais elles sont aussi produites par les leucocytes, les macrophages, les cellules musculaires lisses, les cardiomyocytes et les cellules mésangiales [21].
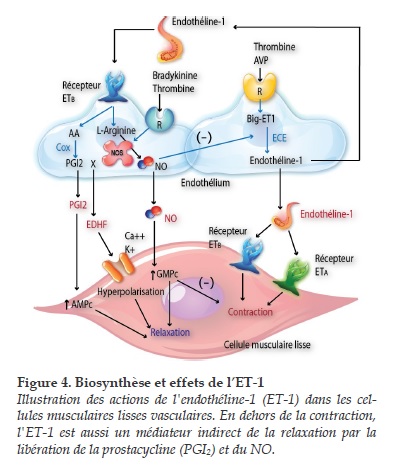
L’ET-1 provient d’un précurseur, le prépro-ET-1 (Figure 3). Après le clivage d’un peptide signal, le prépro-ET-1 est converti en un intermédiaire biologiquement inactif de 38 acides aminés, le Big ET-1, par une enzyme de type convertase. Le Big ET-1 est ensuite clivé au niveau du pont Trp21-Val22 pour donner l’ET-11-21. Cette réaction est catalysée par une enzyme de conversion, l’Endothelin Converting Enzyme-1 (ou ECE-1) dont il existe 4 isoformes (a, b, c et d) dont la traduction protéique s’effectue à partir d’ARN messagers provenant d’épissage alternatif à partir d’un gène unique.
D’autres enzymes, comme les chymases et les métalloprotéases autres que les ECE) peuvent également convertir le Big ET-1 en ET-1. Par ailleurs les chymases clivent le pont Tyr31-Gly32, aboutissant à la synthèse d’un peptide de 31 acides aminés, l’ET-11-31, dont le rôle physiologique est inconnu [22].
L’ET-1 n’est pas stockée dans les vésicules de sécrétion à l’intérieur des cellules endothéliales. Cependant la cascade de réactions aboutissant à la synthèse de l’ET-1 à partir du gène codant son précurseur, le prépro-ET-1 s’effectue en quelques minutes permettant un ajustement très rapide de la production d’ET-1. L’ET-1 est principalement libéré dans le milieu extracellulaire par le pôle basal des cellules endothéliales, en regard des cellules musculaires lisses, suggérant un mécanisme d’action essentiellement paracrine lorsque l’ET-1 se lie aux récepteurs du muscle lisse vasculaire.
Cependant, une action autocrine de l’ET-1 est également possible lorsqu’il active des récepteurs endothéliaux [21]. Sa concentration plasmatique est très faible, de l’ordre de 1-2 pg/ml chez l’adulte sain.
L’ET-1 exerce ses effets en se liant à des récepteurs membranaires dont il existe deux types distincts: le récepteur ET-A et le récepteur ET-B. Tous deux appartiennent à la famille des récepteurs à 7 domaines transmembranaires, couplés aux protéines G. Le récepteur ET-A possède une affinité 100 fois plus grande pour l’ET-1 et l’ET-2 que pour l’ET-3. Le récepteur ET-B possède la même affinité pour les trois isoformes d’endothéline. La liaison de l’endothéline à son récepteur active une protéine G, de type Gq, couplée à la phospholipase C. Cette dernière hydrolyse des phospholipides membranaires, donnant naissance à l’inositol triphosphate d’ une part, et au 1-2 diacylglycerol d’autre part. Par des voies de transduction intracellulaires différentes, ces deux messagers intracellulaires entraînent une augmentation de la concentration intracellulaire de calcium et active la voie de la protéine kinase C favorisant la prolifération cellulaire [22].
Dans le système cardiovasculaire, le récepteur ET-A est exprimé principalement à la surface des cellules musculaires lisses et des cardiomyocytes tandis que le récepteur ET-B est localisé à la fois sur les cellules endothéliales et sur les cellules musculaires lisses [23]. Le récepteur ET-B présent à la surface des cellules endothéliales jouerait un rôle dans la clearance de l’ET-1 plasmatique [24].
Effets cellulaires de l’endothéline
L’ET-1 participe, par son effet vasoconstricteur, à la régulation du tonus vasculaire basal et de la pression artérielle systémique [25]. In vitro, l’ET-1 active la prolifération des cellules musculaires lisses, stimule la synthèse de protéines de la matrice extra-cellulaire, et potentialise les effets du facteur de croissance, TGFβ (Transforming Growth Factor β) et du PDGF (Platelet-Derived Growth Factor) [26]. Son pouvoir mitogénique sur les cellules musculaires lisses est dépendant de la présence respective des récepteurs ET-A et ET-B [27].
L’ET-1 possède également des propriétés pro-inflammatoires [28]. A des concentrations nanomolaires, l’ET-1 possède des effets chronotropes et inotropes [29]. L’ET-1 entraîne la synthèse du NO et de la prostacycline par les cellules endothéliales, via la stimulation des récepteurs endothéliaux de type ET-B [30].
Antagonistes des récepteurs de l’endothéline
Le BQ-123 est un puissant antagoniste sélectif des récepteurs ET-A de l’endothéline. Il s’agit d’un pentapeptide cyclique dérivé d’un autre pentapeptide cyclique, le BE1857A, isolé dans les produits de fermentation de Streptomyces misakiensis [31]. Le BQ-788 est un antagoniste sélectif des récepteurs ET-B de l’endothéline. C’est un tripeptide linéaire dérivé du BQ-123.
Il existe également un antagoniste des récepteurs de l’endothéline capable de discriminer les récepteurs ET-B présents sur la cellule musculaire lisse, de ceux qui sont présents à la surface de l’endothélium. Le PD142893 est un antagoniste aspécifique des récepteurs ET-A et ET-B. Il possède la particularité de pouvoir antagoniser les effets des récepteurs ET-B localisés sur la cellule musculaire lisse, sans bloquer l’action de l’ET-1 sur les récepteurs ET-B endothéliaux [32].
Les progrès dans la compréhension du rôle de l'ET-1 dans l'HTAP ont permis le développement pharmacologique des antagonistes des récepteurs d’ET-1, dont certains, comme le Bosentan (Tracleer®), le Sitaxentan (Thelin®) et l’Ambrisentan (Volibris®) sont actuellement utilisés en clinique [33-35].
Rôle de l’ET-1 dans la physiopathologie de la BPCO
L’endothéline-1 (ET-1), un puissant vasoconstricteur et un agent mitogénique synthétisé par l’endothélium, est aussi un important médiateurs régulant l’activité de la voie Rho-A/Rho-kinases [36, 37]. De nombreuses données expérimentales et cliniques suggèrent que l’ET-1 pourrait jouer un rôle prépondérant dans le remodelage vasculaire lié au tabagisme.
Goerre et al. ont montré que le tabac entraîne une élévation transitoire de la concentration plasmatique d’ET-1 dans la circulation systémique [38].
L’exposition en aiguë d’animaux à la fumée de tabac stimule la prolifération des cellules vasculaires pulmonaires et bronchiques [39]. A l’inverse, la prolifération cellulaire est réduite par l’inhibition de l’effet de l’ET-1 avec des antagonistes des récepteurs ET-A [40]. De plus, l’exposition chronique à la fumée de tabac augmente de façon durable la transcription du gène codant l’ET-1 par les cellules vasculaires pulmonaires [41]. Enfin, la concentration d’ET-1 tant dans la circulation systémique que dans le liquide broncho-alvéolaire est augmentée chez les patients BPCO [42].
CONCLUSION
Le dysfonctionnement endothélial est non seulement présent chez les malades avec une BPCO mais aussi chez les fumeurs sans trouble ventilatoire obstructif. Ce dysfonctionnement pourrait être lié à la fois la diminution de la production du No et l’augmentation de l’activité du système d’ET-1.
Ce dysfonctionnement souligne l'importance et la précocité des effets délétères du tabac dont l'effet cytotoxique semble de toute évidence précéder de très loin les altérations fonctionnelles respiratoires et gazométriques des patients atteints de BPCO post-tabagiques.
CONFLIT D’INTERETS
Aucun.
REFERENCES
1. Lopez AD, Shibuya K, Rao C, Mathers CD, Hansell AL, Held LS, Schmid V, Buist S. Chronic obstructive pulmonary disease : current burden and future projections. Eur Respir J 2006; 27:397-412.
2. Murray CJ, Lopez AD. Alternative projections of mortality and disability by cause 1990-2020: Global burden of disease study. Lancet 1997; 349:1498-1504.
3. Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med. 1995; 333(4):214-21
4. Hoffman D, Wynder EL. Chemical constituents and bioactivity of tobacco smoke. In Tobacco, a Major International Health Hazard. D.G. Zaridge and R.Peto, editors. Oxford University Press, London. 1986; 145-165.
5. Powell JT, Higman DJ. Smoking, nitric oxide and the endothelium. Br J Surg 1994; 81(6):785-7.
6. Kiowski W, Linder L, Stoschitzky K, Pfisterer M, Burckhardt D, Burkart F, Buhler FR. Diminished vascular response to inhibition of endothelium-derived nitric oxide and enhanced vasoconstriction to exogenously administered endothelin-1 in clinically healthy smokers. Circulation 1994; 90(1):27-34.
7. Naeije R. Pulmonary hypertension and right heart failure in chronic obstructive pulmonary disease. The Proceedings of the American Thoracic Society 2005; 2:20-2.
8. Barberà JA, Peinado VI, Santos S. Pulmonary hypertension in chronic obstructive pulmonary disease. Eur Respir J 2003; 21:892-905.
9. Peinado VI, Barberà JA, Ramirez J, Gomez FP, Roca J, Jover L, Gimferrer JM, Rodriguez-Roisin R. Endothelial dysfunction in pulmonary arteries of patients with mild COPD. Am J Physiol. 1998; 274: L908–L103.
10. Santos S, Peinado VI, Ramirez J, Melgosa T, Roca J, Rodriguez-Roisin R, Barbera JA. Characterization of pulmonary vascular remodeling in smokers and patients with mild COPD. Eur Respir J 2002; 19: 632–638.
11. MacNee W. Pulmonary and systemic oxidant/ antioxidant imbalance in chronic obstructive pulmonary disease. Proc Am Thorac Soc 2005; 2:50-60.
12. Weitzenblum E, Hirth C, Ducolone A, Mirhom R, Rasaholinjanahary J, Ehrhart M. Prognostic value of pulmonary artery pressure in chronic obstructive pulmonary disease.Thorax 1981; 36(10):752-8.
13. Magee F, Wright JL, Wiggs BR, Pare PD, Hogg JC. Pulmonary vascular structure and function in chronic obstructive pulmonary disease. Thorax 1988;43 (3) :183-9.
14. Peinado VI, Barbera JA, Abate P, Ramirez J, Roca J, Santos S, Rodriguez-Roisin R.Inflammatory reaction in pulmonary muscular arteries of patients with mild chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1999; 159(5 Pt 1):1605-11.
15. Griendling KK, Alexander RW. Endothelial control of the cardiovascular system: recent advances. FASEB J. 1996; 10(2):283-92.
16. Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980 ; 288(5789):373-6.
17. Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med 1993; 329(27):2002-12.
18. Su Y, Han W, Giraldo C, De Li Y, Block ER. Effect of cigarette smoke extract on nitric oxide synthase in pulmonary artery endothelial cells. Am J Respir Cell Mol Biol 1998; 19(5):819-25.
19. Robbins RA, Barnes PJ, Springall DR, Warren JB, Kwon OJ, Buttery LD, Wilson AJ, Geller DA, Polak JM. Expression of inducible nitric oxide in human lung epithelial cells. Biochem Biophys Res Commun 1994; 30:20918.
20. Keating VM, Collins PD, Scott DM, Barnes PJ. Differences in interleukin-8 and tumor necrosis factor-a in niduced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med 1996; 153:530-34.
21. Okogbule-Wonodi AC, Ibe BO, Yue BW, Hsu S, Raj JU. Phosphodiesterase activity in intrapulmonary arteries and veins of perinatal lambs. Mol Genet Metab 1998; 65 (3):229-37.
22. Hanson KA, Ziegler JW, Rybalkin SD, Miller JW, Abman SH, Clarke WR. Chronic pulmonary hypertension increases fetal lung cGMP phosphodiesterase activity. Am J Physiol 1998; 275(5 Pt 1):L931-41.
23. Black SM, Sanchez LS, Mata-Greennwood E, Bekker JM, Steinhorm RH, Fineman JR. sGC and PDE 5 are elevated in lambs with increased pulmonary blood flow and pulmonary hypertension. Am J Physiol 2001; 281: L1051-L1057.
24. Ziegler JW, Ivy DD, Fox JJ, Kinsella JP, Clarke WR, Abman SH. Dipyridamole, a cGMP phosphodiesterase inhibitor, causes pulmonary vasodilation in the ovine fetus. Am J Physiol 1995; 269 (Heart Circ Physiol): H473H479.
25. Thusu KG, Morin FC, Russel JA, Steinhorn RH. The cGMP phosphodiesterase inhibitor Zaprinast enhances the effect of nitric oxide. Am J Respir Crit Care Med 1995; 152: 1605-1610.
26. Hanson KA, Burns F, Rybalkin SD, Miller JW, Beavo J, Clarke WR. Developmental changes in lung cGMP phosphodiesterase-5 activity, protein and message. Am J Respir Crit Care Med 1998; 158: 279-288.
27. Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002 ; 346 : 896–903.
28. Jozsef L, Khreiss T, Fournier A, Chan JS and Filep JG, Extracellular signal-regulated kinase plays an essential role in endothelin-1-induced homotypic adhesion of human neutrophil granulocytes. Br. J. Pharmacol. 2002; pp. 1167–1174.
29. Ishikawa T, Yanagisawa M, Kimura S, Goto K and Masaki T, Positive inotropic action of novel vasoconstrictor peptide endothelin on guinea pig atria. Am. J. Physiol 1988; pp. H970–H973.
30. Hirata Y, Emori T, Eguchi S et al., Endothelin receptor subtype B mediates synthesis of nitric oxide by cultured bovine endothelial cells. J. Clin. Invest 1993; pp. 1367–1373.
31. Ihara M, Noguchi K, Saeki T, Fukuroda T, Tsuchida S, Kimura S, Fukami T, Ishikawa K, Nishikibe M, Yano M. Biological profiles of highly potent novel endothelin antagonists selective for the ETA receptor. Life Sci 1992; pp. 247–255.
32. SA Douglas. Novel receptor antagonists welcome a new era in endothelin biology. Trends Pharmacol. Sci 1994; pp. 313–316.
33. Channick RN, Simonneau G, Sitbon O, Robbins IM, Frost A, Tapson VF, Badesch DB, Roux S, Rainisio M, Bodin F, Rubin LJ. Effects of the dual endothelinreceptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study. Lancet 2001; 358 : 1119–1123.
34. Hanson KA, Ziegler JW, Rybalkin SD, Miller JW, Abman SH, Clarke WR. Chronic pulmonary hypertension increases fetal lung cGMP phosphodiesterase activity. Am J Physiol 1998; 275(5 Pt 1):L931-41.
35. Barst RJ, Langleben D, Frost A, Horn EM, Oudiz R, Shapiro S, McLaughlin V, Hill N, Tapson VF, Robbins IM, Zwicke D, Duncan B, Dixon RA, Frumkin LR; STRIDE-1 Study Group. Sitaxsentan Therapy for Pulmonary Arterial Hypertension. Am J Respir Crit Care Med 2004 ; 169 : 441-7.
36. Pollock DM, Keith TL and Highsmith RF, Endothelin receptors and calcium signaling. FASEB 1995, 1196–204
37. Ohlstein EH, Arleth A, Bryan H, Elliott JD and Sung CP. The selective endothelin ETA receptor antagonist BQ123 antagonizes endothelin-1-mediated mitogenesis. Eur. J. Pharmacol. 225. 1992, pp. 347–350.
38. Goerre S, Staehli C, Shaw S, Luscher TF. Effect of cigarette smoking and nicotine on plasma endothelin-1 levels. J Cardiovasc Pharmacol. 1995; 26 Suppl 3:S236-8.
39. Sekhon HS, Wright JL and Churg A.Cigarette smoke causes rapid cell proliferation in small airways and associated pulmonary arteries. Am J Physiol. 1994; 267:L557-63.
40. Dadmanesh F and Wright JL. Endothelin-A receptor antagonist BQ-610 blocks cigarette smoke-induced mitogenesis in rat airways and vessels. Am J Physiol. 1997; 272(4 Pt 1):L614-8.
41. Wright JL, Tai H and Churg H. Cigarette smoke induces persisting increases of vasoactive mediators in pulmonary arteries.Am J Respir Cell Mol Biol. 2004; 31 (5):501-9.
42. Bacakoğlu F, Atasever A, Ozhan et al. Plasma and bronchoalveolar lavage fluid levels of endothelin-1 in patients with chronic obstructive pulmonary disease and pulmonary hypertension. Respiration 2003;70:594-9.
43. Duong-Quy S, Dao P, Hua-Huy T, Guilluy C et al. Increased Rho-kinase expression and activity and pulmonary endothelial dysfunction in smokers with normal lung function. Eur Respir J 2011;37(2):349-55.

FIGURES
REFERENCES
1. Lopez AD, Shibuya K, Rao C, Mathers CD, Hansell AL, Held LS, Schmid V, Buist S. Chronic obstructive pulmonary disease : current burden and future projections. Eur Respir J 2006; 27:397-412.
2. Murray CJ, Lopez AD. Alternative projections of mortality and disability by cause 1990-2020: Global burden of disease study. Lancet 1997; 349:1498-1504.
3. Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med. 1995; 333(4):214-21
4. Hoffman D, Wynder EL. Chemical constituents and bioactivity of tobacco smoke. In Tobacco, a Major International Health Hazard. D.G. Zaridge and R.Peto, editors. Oxford University Press, London. 1986; 145-165.
5. Powell JT, Higman DJ. Smoking, nitric oxide and the endothelium. Br J Surg 1994; 81(6):785-7.
6. Kiowski W, Linder L, Stoschitzky K, Pfisterer M, Burckhardt D, Burkart F, Buhler FR. Diminished vascular response to inhibition of endothelium-derived nitric oxide and enhanced vasoconstriction to exogenously administered endothelin-1 in clinically healthy smokers. Circulation 1994; 90(1):27-34.
7. Naeije R. Pulmonary hypertension and right heart failure in chronic obstructive pulmonary disease. The Proceedings of the American Thoracic Society 2005; 2:20-2.
8. Barberà JA, Peinado VI, Santos S. Pulmonary hypertension in chronic obstructive pulmonary disease. Eur Respir J 2003; 21:892-905.
9. Peinado VI, Barberà JA, Ramirez J, Gomez FP, Roca J, Jover L, Gimferrer JM, Rodriguez-Roisin R. Endothelial dysfunction in pulmonary arteries of patients with mild COPD. Am J Physiol. 1998; 274: L908–L103.
10. Santos S, Peinado VI, Ramirez J, Melgosa T, Roca J, Rodriguez-Roisin R, Barbera JA. Characterization of pulmonary vascular remodeling in smokers and patients with mild COPD. Eur Respir J 2002; 19: 632–638.
11. MacNee W. Pulmonary and systemic oxidant/ antioxidant imbalance in chronic obstructive pulmonary disease. Proc Am Thorac Soc 2005; 2:50-60.
12. Weitzenblum E, Hirth C, Ducolone A, Mirhom R, Rasaholinjanahary J, Ehrhart M. Prognostic value of pulmonary artery pressure in chronic obstructive pulmonary disease.Thorax 1981; 36(10):752-8.
13. Magee F, Wright JL, Wiggs BR, Pare PD, Hogg JC. Pulmonary vascular structure and function in chronic obstructive pulmonary disease. Thorax 1988;43 (3) :183-9.
14. Peinado VI, Barbera JA, Abate P, Ramirez J, Roca J, Santos S, Rodriguez-Roisin R.Inflammatory reaction in pulmonary muscular arteries of patients with mild chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1999; 159(5 Pt 1):1605-11.
15. Griendling KK, Alexander RW. Endothelial control of the cardiovascular system: recent advances. FASEB J. 1996; 10(2):283-92.
16. Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980 ; 288(5789):373-6.
17. Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med 1993; 329(27):2002-12.
18. Su Y, Han W, Giraldo C, De Li Y, Block ER. Effect of cigarette smoke extract on nitric oxide synthase in pulmonary artery endothelial cells. Am J Respir Cell Mol Biol 1998; 19(5):819-25.
19. Robbins RA, Barnes PJ, Springall DR, Warren JB, Kwon OJ, Buttery LD, Wilson AJ, Geller DA, Polak JM. Expression of inducible nitric oxide in human lung epithelial cells. Biochem Biophys Res Commun 1994; 30:20918.
20. Keating VM, Collins PD, Scott DM, Barnes PJ. Differences in interleukin-8 and tumor necrosis factor-a in niduced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med 1996; 153:530-34.
21. Okogbule-Wonodi AC, Ibe BO, Yue BW, Hsu S, Raj JU. Phosphodiesterase activity in intrapulmonary arteries and veins of perinatal lambs. Mol Genet Metab 1998; 65 (3):229-37.
22. Hanson KA, Ziegler JW, Rybalkin SD, Miller JW, Abman SH, Clarke WR. Chronic pulmonary hypertension increases fetal lung cGMP phosphodiesterase activity. Am J Physiol 1998; 275(5 Pt 1):L931-41.
23. Black SM, Sanchez LS, Mata-Greennwood E, Bekker JM, Steinhorm RH, Fineman JR. sGC and PDE 5 are elevated in lambs with increased pulmonary blood flow and pulmonary hypertension. Am J Physiol 2001; 281: L1051-L1057.
24. Ziegler JW, Ivy DD, Fox JJ, Kinsella JP, Clarke WR, Abman SH. Dipyridamole, a cGMP phosphodiesterase inhibitor, causes pulmonary vasodilation in the ovine fetus. Am J Physiol 1995; 269 (Heart Circ Physiol): H473H479.
25. Thusu KG, Morin FC, Russel JA, Steinhorn RH. The cGMP phosphodiesterase inhibitor Zaprinast enhances the effect of nitric oxide. Am J Respir Crit Care Med 1995; 152: 1605-1610.
26. Hanson KA, Burns F, Rybalkin SD, Miller JW, Beavo J, Clarke WR. Developmental changes in lung cGMP phosphodiesterase-5 activity, protein and message. Am J Respir Crit Care Med 1998; 158: 279-288.
27. Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002 ; 346 : 896–903.
28. Jozsef L, Khreiss T, Fournier A, Chan JS and Filep JG, Extracellular signal-regulated kinase plays an essential role in endothelin-1-induced homotypic adhesion of human neutrophil granulocytes. Br. J. Pharmacol. 2002; pp. 1167–1174.
29. Ishikawa T, Yanagisawa M, Kimura S, Goto K and Masaki T, Positive inotropic action of novel vasoconstrictor peptide endothelin on guinea pig atria. Am. J. Physiol 1988; pp. H970–H973.
30. Hirata Y, Emori T, Eguchi S et al., Endothelin receptor subtype B mediates synthesis of nitric oxide by cultured bovine endothelial cells. J. Clin. Invest 1993; pp. 1367–1373.
31. Ihara M, Noguchi K, Saeki T, Fukuroda T, Tsuchida S, Kimura S, Fukami T, Ishikawa K, Nishikibe M, Yano M. Biological profiles of highly potent novel endothelin antagonists selective for the ETA receptor. Life Sci 1992; pp. 247–255.
32. SA Douglas. Novel receptor antagonists welcome a new era in endothelin biology. Trends Pharmacol. Sci 1994; pp. 313–316.
33. Channick RN, Simonneau G, Sitbon O, Robbins IM, Frost A, Tapson VF, Badesch DB, Roux S, Rainisio M, Bodin F, Rubin LJ. Effects of the dual endothelinreceptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study. Lancet 2001; 358 : 1119–1123.
34. Hanson KA, Ziegler JW, Rybalkin SD, Miller JW, Abman SH, Clarke WR. Chronic pulmonary hypertension increases fetal lung cGMP phosphodiesterase activity. Am J Physiol 1998; 275(5 Pt 1):L931-41.
35. Barst RJ, Langleben D, Frost A, Horn EM, Oudiz R, Shapiro S, McLaughlin V, Hill N, Tapson VF, Robbins IM, Zwicke D, Duncan B, Dixon RA, Frumkin LR; STRIDE-1 Study Group. Sitaxsentan Therapy for Pulmonary Arterial Hypertension. Am J Respir Crit Care Med 2004 ; 169 : 441-7.
36. Pollock DM, Keith TL and Highsmith RF, Endothelin receptors and calcium signaling. FASEB 1995, 1196–204
37. Ohlstein EH, Arleth A, Bryan H, Elliott JD and Sung CP. The selective endothelin ETA receptor antagonist BQ123 antagonizes endothelin-1-mediated mitogenesis. Eur. J. Pharmacol. 225. 1992, pp. 347–350.
38. Goerre S, Staehli C, Shaw S, Luscher TF. Effect of cigarette smoking and nicotine on plasma endothelin-1 levels. J Cardiovasc Pharmacol. 1995; 26 Suppl 3:S236-8.
39. Sekhon HS, Wright JL and Churg A.Cigarette smoke causes rapid cell proliferation in small airways and associated pulmonary arteries. Am J Physiol. 1994; 267:L557-63.
40. Dadmanesh F and Wright JL. Endothelin-A receptor antagonist BQ-610 blocks cigarette smoke-induced mitogenesis in rat airways and vessels. Am J Physiol. 1997; 272(4 Pt 1):L614-8.
41. Wright JL, Tai H and Churg H. Cigarette smoke induces persisting increases of vasoactive mediators in pulmonary arteries.Am J Respir Cell Mol Biol. 2004; 31 (5):501-9.
42. Bacakoğlu F, Atasever A, Ozhan et al. Plasma and bronchoalveolar lavage fluid levels of endothelin-1 in patients with chronic obstructive pulmonary disease and pulmonary hypertension. Respiration 2003;70:594-9.
43. Duong-Quy S, Dao P, Hua-Huy T, Guilluy C et al. Increased Rho-kinase expression and activity and pulmonary endothelial dysfunction in smokers with normal lung function. Eur Respir J 2011;37(2):349-55.
ARTICLE INFO
DOI: 10.12699/jfvp.2.5.2011.21
Conflict of Interest
Non
Date of manuscript receiving
9/3/2011
Date of publication after correction
15/9/2011
Article citation
Duong-Quy S, Le-Dong N.N, Hua-Huy T, Dinh-Xuan A.T. Endothelial dysfunction in COPD: Role of nitric oxide and endothelin-1. J Func Vent Pulm 2011;02(05):21-28.



Copyright: jfvpulm.com