
 English
English
 French
French
 Pubmed
Pubmed Google Scholar
Google Scholar Cross Ref
Cross Ref Visistor: 785
Visistor: 785Nouvelle définition et classification de l’hypertension pulmonaire
New definition and classification of Pulmonary Hypertension
S. Duong-Quya b, N.N. Le-Donga, C. Duong-Ngoa, B. Mai Huu Thanhb, T. Hua-Huya, A.T. Dinh-Xuana
a Hôpital Cochin & Faculté de Médecine René-Descartes, Paris, France
b Collège de Médecine Lam Dong, Da Lat, Viet Nam
Corresponding author
Dr Sy Duong-Quy
Laboratoire UPRES-2511, Faculté de Médecine René-Descartes
Services des EFR, Hôpital Cochin, Paris - France
Tel : +33. 0158413065; Adresse e-mail: sy.duong-quy@cch.aphp
DOI: 10.12699/jfvp.1.1.2010.35
ABSTRACT
Many advances in diagnosis and treatment of pulmonary hypertension have been considerably obtained in recent years due to a better understanding of pathogenesis of the disease. In the classification of pulmonary hypertension, the etiology of the disease is diverse. Hence, it would be preferable to refer the patients to specialist centers earlier to perform the whole diagnosis including right heart catheterization. However, before the proceeding of catheterization to confirm the diagnosis, the screening and early diagnosis of the disease are necessary. Given the complexity in pathogenesis of pulmonary hypertension, the multidisciplinary management is still required.
Key-words: Pulmonary hypertension, Pulmonary arterial pressure, Plexiform lessions
RÉSUMÉ
Des progrès considérables dans le diagnostic et le traitement de l’HTP ont été obtenus ces dernières années, grâce à une meilleure compréhension de la pathogénie de la maladie. Dans la classification de l’HTP, les étiologies de la maladie sont très diverses. Ainsi, il est préférable d’adresser précocement les patients vers des centres spécialisés pour une évaluation diagnostique complète qui inclut la réalisation d’un cathétérisme cardiaque droit. Cependant, avant de procéder au cathétérisme pour le diagnostic de certitude, le dépistage et le diagnostic précoce de l’HTP sont nécessaires. Etant donné la nature complexe de la pathogénie de l’HTAP, une prise en charge multidisciplinaire reste indispensable.
Mots-clés: Hypertension pulmonaire, Pression artérielle pulmonaire, Lésions plexiformes
SYNTHÈSE DE LA PHYSIOPATHOLOGIE ET PATHOLOGIE MOLÉCULAIRE DE L’HTP
Physiopathologie de l’HTP
Le principal mécanisme décrit est celui de la vasoconstriction pulmonaire induite par l’hypoxie alvéolaire [4]. La vasoconstriction est due à un déséquilibre de la balance entre les vasodilatateurs (monoxyde d’azote, prostacycline) et les vasoconstricteurs (thromboxane A2, endothéline 1, angiotensine II), médié par l’hypoxie.
Le deuxième mécanisme physiopathologique impliqué dans l’HTP est lié à la diminution de la surface de section transversale du lit vasculaire pulmonaire [5]. Celle-ci est généralement due à des du parenchyme pulmonaire (fibrose pulmonaire, sclérodermie systémique) ou l’obstruction thromboembolique des artères pulmonaires proximales ou distales.
L’autre mécanisme occupant un rôle non négligeable dans la physiopathologie de l’HTP est la surcharge de volume et de pression dans les maladies cardiovasculaires. Cette surcharge est causée par les shunts intracardiaques qui augmentent le volume de remplissage du cœur droit, et par l’élévation, en amont, des pressions veineuse et artérielle pulmonaires. L’élévation des pressions veineuses et artérielles pulmonaires est principalement observée dans la maladie veino-occlusive pulmonaire et l’hémangiomatose capillaire pulmonaire [6, 7].
Pathologie biomoléculaire de l’HTP
La pathologie biomoléculaire de l’HTP est marquée par un déséquilibre entre les vasoconstricteurs et les vasodilatateurs entraînant un trouble du tonus vasculaire pulmonaire. C’est pourquoi le rôle des médiateurs vasoconstricteurs et vasodilatateurs est primordiale dans le contrôle du tonus vasculaire pulmonaire.
Endothéline-1 (ET-1)
L’ET-1 est un vasoconstricteur puissant possédant également des effets mitogéniques. La concentration plasmatique d’ET-1 est corrélée à la résistance vasculaire pulmonaire et à la survie des patients atteints d’HTP [8, 9]. La liaison de l'endothéline à son récepteur ETA et ETB active une protéine G couplée à la phospholipase C entraînant une augmentation de la concentration intracellulaire de calcium responsable de la contraction du muscle lisse vasculaire.
Sérotonine (5-hyroxytryptamine, 5-HT)
L’activité du système 5-HT est augmentée dans l’HTTP. Les transporteurs de la sérotonine (SERT) et du tryptophane hydroxylase 1 (TPH1) sont impliqués dans la synthèse de 5-HT [10, 11]. Les anorexigènes (aminorex et fenfluramine) provoquent une HTP en agissant comme substrats du SERT, induisant ainsi la libération de 5-HT [12].
Monoxyde d’azote (NO)
Le NO est synthétisé par l’enzyme NO synthase en présence ses cofacteurs. Le NO active le guanylate cyclase soluble (sGC) qui catalyse la formation de guanosine monophosphate (cGMP) et induit la vasodilatation via la protéine kinase G (PKG). Le NO inhible également la prolifération des cellules musculaires lisses (CML) et a un effet antiagrégant plaquettaire [13]. Dans l’HTP, l’expression de la NOS endothéliale, la production de NO et la biodisponibilité du NO sont diminuées dans les tissus pulmonaires [14].
La Prostacyclin (PGI2) et le Thromboxane-A2 (TxA2)
La PGI2 est un vasodilatateur endogène agissant sur la synthèse de l’adénosine monophosphate cyclique (cAMP). Dans l’HTP, l’expression de la PGI2 et de la prostacycline synthase est diminuée dans les artères pulmonaires, en particulier dans l’HTAP avec lésion plexiforme [15, 16].
En revanche, le TxA2, un puissant agrégeant plaquettaire, est aussi un important vasoconstricteur de la voie cyclo-oxygénase. L’expression des récepteurs du TxA2 et son métabolisme est élevés chez les patients atteints d’HTP idiopathique [17, 18].
Le Vasoactive intestinal peptide (VIP)
Connu comme médiateur neuroendocrine et vasodilatateur systémique, le VIP a un rôle prépondérant dans la pathologie de l’HTP [19]. En effet, une diminution de l’expression de VIP dans les tissus pulmonaires et de la concentration sérique a été retrouvée chez les patients avec l’HTP[20]. Le VIP serait une cible thérapeutique intéressante dans le futur pour l’HTP.
ACTUALISATION DE LA DÉFINITION ET CLASSIFICATION DE L’HTTP
Définition de l’HTTP
La définition hémodynamique actuelle de l’HTP est une PAPm ≥ 25 mmHg au repos mesurée par le cathétérisme droit, associée à une pression artérielle pulmonaire occluse ≤ 15 mmHg (HTP pré-capillaire) ou > 15 mmHg (HTP post-capilaire) [2, 3]. Les valeurs normales de la pression artérielle pulmonaire moyenne (PAPm) sont de l’ordre de 14,3 ± 3,3 mmHg chez les sujets sains [21]. L’ancienne définition de l’HTP basée sur la PAPm > 30 mmHg à l’effort n’est plus utilisée dans les nouvelles recommandations en raison des données scientifiques insuffisantes.
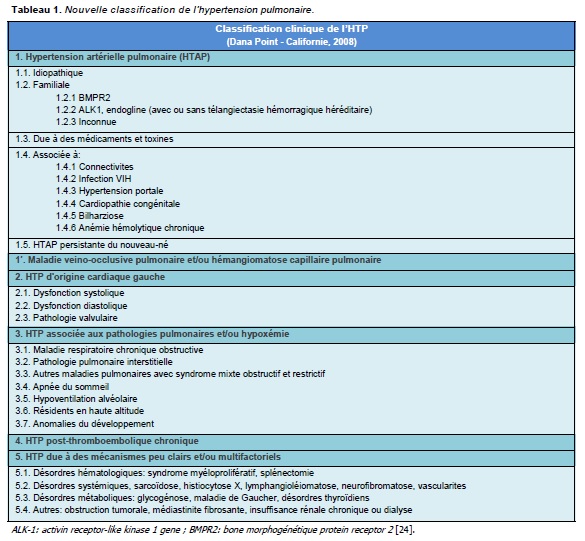
Classification de l’HTP
La nouvelle classification de l’HTP est présentée sur le Tableau 1. Elle reprend le plan la classification de 1998 (Evian), mise à jour en 2003 (Venise) et modifié en 2008 (Dana Point) avec des points spécifiques.
Groupe 1 (hypertension artérielle pulmonaire, HTAP)
Dans la nouvelle classification, l’HTAP familiale est désormais remplacée par l’HTAP héritable car des mutations sur les gènes codant pour BMPR2, ALK1 ou endoglin ont été mises en évidence chez des patients atteints d’HTAP idiopathique (HTAPi) sporadique.
L’HTAP due à des médicaments et toxines survient en association avec l’exposition aux anorexigènes (fenfluramine, dexfenfluramine, aminorex), aux psychostimulants (amphétamine, méta-amphétamines) ou à la cocaïne.
L’HTAP liée aux cardiopathies congénitales a été actualisée en incluant les différents aspects cliniques et anatomo-pathophysiologiques spécifiques de chaque patient.
L’HTAP associée aux autres pathologies ont les mêmes caractéristiques cliniques que l’HTAPi avec lésions vasculaires plexiformes. Dans ce groupe, l’HTAP associée à la bilharziose et aux anémies hémolytiques a été ajoutée. La première est liée à l’hypertension portale et la deuxième à la diminution de l’activité de la voie GMPc due à une consommation élevée en NO.
Groupe 1’
La maladie veino-occlusive pulmonaire et l’hémangiomatose capillaire appartiennent à ce sous-groupe. Ces maladies partagent certaines caractéristiques de l’HTAPi mais possèdent des différences dans la prise en charge [22].
Groupe 2 et groupe 3
Le groupe 2 inclut les cardiopathies gauches comprenant les dysfonctions ventriculaires et les pathologies valvulaires.
Le groupe 3 inclut les maladies respiratoires chroniques avec ou sans l’hypoxémie telles que la BPCO, le syndrome des apnées du sommeil, l’hypoventilation alvéolaire.
Groupe 4
Il comprend l’HTP post emboliques chroniques. La distinction entre forme proximale et distale de cette pathologie a été supprimée à cause de l’absence de critères précis permettant de différencier ces deux formes [23].
Groupe 5
Ce groupe inclut des pathologies diverses conduisant à une HTP dont les mécanismes pathogéniques n’ont pas encore été bien élucidés.
DIAGNOSTIC DE L’HTP
Diagnostic précoce
Le diagnostic de l’HTAP peut être difficile au stade précoce du fait du caractère asymptomatique de la maladie à ce stade et des symptômes initiaux aspécifiques. Malgré ces défis, l’identification des patients est possible grâce à la classification fonctionnelle NYHA (New York Heart Association) est possible. Le diagnostic précoce de l’HTP repose sur le dépistage des groupes à haut risque.
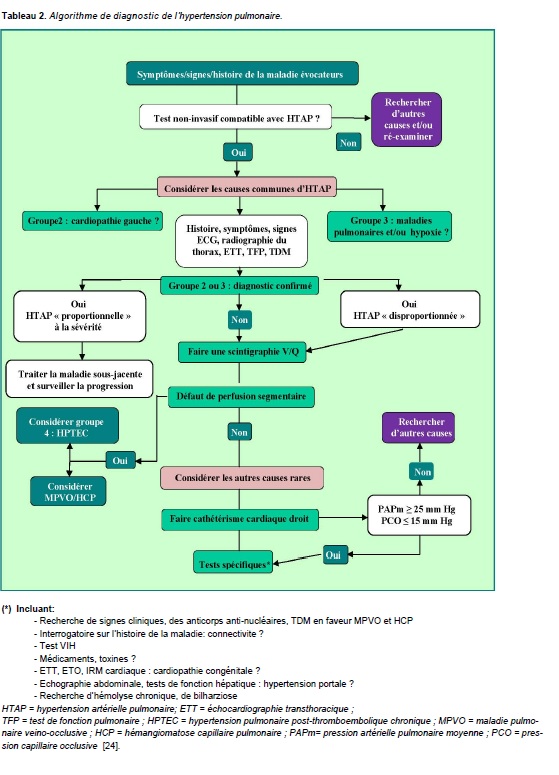
Algorithme diagnostique (Tableau 2)
Selon la classification actuelle, les étiologies de l’HTAP sont nombreuses, incluant les formes idiopathique, héréditaire, induites par des médicaments et des toxiques ou associées à différentes pathologies (maladies respiratoires, connectivite, infection VIH). D’où le besoin d’adresser précocement les patients à des centres spécialisés pour une évaluation diagnostique complète qui inclut la réalisation d’un cathétérisme droit, examen indispensable pour le diagnostic de l’HTAP. Le dépistage et la prise en charge précoces sont d’autant plus importants que les données ont montré que les patients HTAP de classe II NYHA avaient un meilleur pronostic et une survie améliorée par rapport aux patients de classe III [25].
Diagnostic de l’HTAP (groupe 1)
Présentation clinique
Les signes cliniques de l’HTAP sont très variés et non spécifiques, liés au retentissement de la maladie sur le cœur droit (cœur pulmonaire chronique) ou à la maladie sous-jacente, comme la télangiectasie, l’ulcère digitale et la sclérodactie dans la sclérodermie systémique. Les antécédents en faveur de l’HTP et les facteurs de risques de la maladie doivent être recherchés.
Le diagnostique clinique probable d’HTAP repose donc sur la présence des signes cliniques évocateurs face à un tableau clinique suspect. La dyspnée d’effort est retrouvée dans la plupart des cas
(95%). Des douleurs thoraciques, des lipothymies ou des syncopes surviennent souvent à l’effort, suggérant la gravité de la maladie. Des hémoptysies de faible quantité peuvent survenir au cours de l’HTAP.
Les signes d’insuffisance ventriculaire droite doivent être recherchés systématique. L'auscultation cardiaque peut retrouver un éclat de B2 au foyer pulmonaire, un souffle systolique d'insuffisance tricuspide, et parfois un souffle diastolique d'insuffisance pulmonaire.
Examens complémentaires
Pou confirmer le diagnostic de l’HTAP, plusieurs examens complémentaires sont nécessaires.
- Radiographie et scanner thoracique
La radiographie thoracique retrouve une hypertrophie du tronc et des branches proximales des artères pulmonaires avec augmentation de l’index cardio-thoracique (coeur pulmonaire chronique). Elle permet aussi de retrouver des lésions parenchymateuses associées. Cependant, ces anomalies sont mieux caractérisées par le scanner thoracique.
- Electrocardiogramme (ECG)
L’ECG permet de détecter des signes d’hypertrophie au niveau auriculaire droite (onde P ample en DII-DIII et bifide en V1) et ventriculaire droite (aspect S1Q3 lié à la dextrorotation, grande onde R en V1 et R < S en V6, troubles de la repolarisation sur les précordiales droites).
- Echocardiographie trans-thoracique (ETT)
L’ETT couplé au Doppler est l’examen de référence lorsque suspicion d’HTP. Elle permet de détecter l’HTAP grâce aux différents paramètres mesurés (vitesse maximale de la fuite tricuspidienne, PAP systolique, pression oreillette droite). Elle permet d’évaluer la dysfonction ventriculaire gauche et de rechercher une valvulopathie. Cet examen permet aussi de rechercher une cardiopathie congénitale ou un shunt droit-gauche par ouverture du foramen ovale. Une échographie cardiaque trans-oesophagienne est parfois nécessaire pour la détection des anomalies cardiaques, en particulier au niveau du septum auriculaire.
- Cathétérisme cardiaque droit
Le cathétérisme cardiaque droit est l’examen de référence indispensable pour confirmer le diagnostic d’HTAP. Il permet aussi d’évaluer la sévérité de la maladie et de réaliser au cours de l’examen un test de vasoréactivité du NO (monoxyde d’azote) à la dose de 10 - 20 ppm pendant 5 minutes par l’intermédiaire d’un masque facial ou d’un embout buccal. Un test positif est défini par une diminution de la PAPm ≥ 10 mmHg pour atteindre une valeur ≤ 40 mmHg avec un débit cardiaque augmenté ou inchangé [25].
- Marqueurs biochimiques
Les marqueurs biochimiques sont un des examens importants dans le pronostic et le suivi de la maladie. Un taux élevé d’acide urique dans le sérum est associé à un mauvais pronostic chez les patients atteints d’HTAPi [26]. Une élévation des taux circulants des peptides natriurétiques (BNP, NT-proBNP) est associée à un mauvais pronostic au cours de l’HTAPi ou de l’HTAP associée à une sclérodermie [27-29].
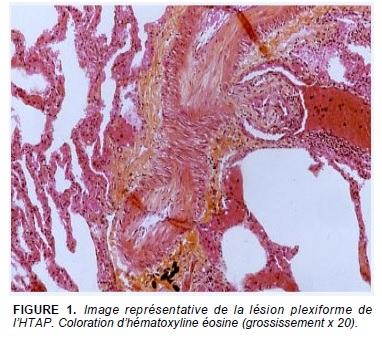
Lésions caractéristiques de l’HTAP
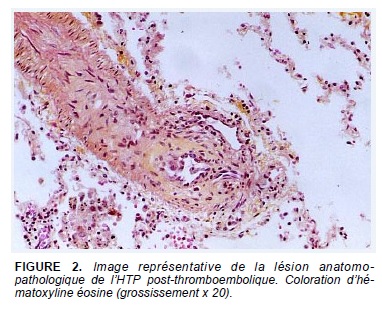
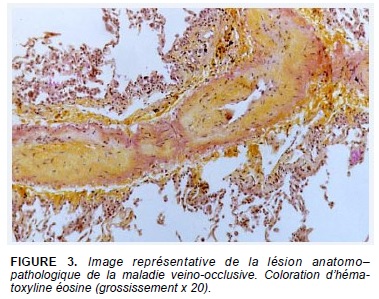
La lésion histologique de l’HTAP est une artériopathie plexiforme (Fig. 1) qui n’a pas été vue dans les autres types de l’HTP et dans l’HTP post-thromboembolique chronique. Cette lésion est caractérisée par une hypertrophie de la média liée à une prolifération excessive des cellules musculaires lisses et à l’infiltration de matrice conjonctive et élastine, par une fibrose de l’intima lié à l’infiltration des fibroblastes, des myofibroblastes et des cellules musculaires lisses. Elle est souvent associée à des lésions plexiformes et des thromboses organisées et recanalisées. Cependant, les lésions vasculaires dans l’HTAP peuvent être associées ou isolées chez le même patient et entre différents patients (Fig.2 et 3).
Dépistage de l’HTP
Les patients à risque pour l’HTP sont soumis à un dépistage périodique qui comprend une recherche de la mutation du gène BMPR2, d’une sclérodermie systémique, ou dune hypertension portale. L’examen complémentaire le plus approprié chez des patients suspectés d’avoir une HTP, avec interrogatoire, examen clinique, radiographie de thorax et électrocardiogramme (ECG)
réalisés au préalable, est l’échographie cardiaque. L’évaluation des autres étiologies possibles, telles que les maladies thrombo-emboliques, est recommandée pour tous les patients suspectés d'avoir une HTP. Enfin, le diagnostic de l'HTAP nécessite une confirmation par le cathétérisme cardiaque droit.
Les nouveautés dans le diagnostic de l’HTP
Il est bien établi que les patients atteints d’HTP présentent des symptômes qui sont liés à des troubles hémodynamiques en rapport avec la maladie vasculaire pulmonaire sous-jacente. Les symptômes de l’HTP sont aspécifiques et apparaissent typiquement à l’effort incluant dyspnée, asthénie, syncope, angine de poitrine et toux et. La rétention liquidienne, en particulier l’ascite, apparaît dans les cas les plus sévères.
Si l’examen clinique peut mettre en évidence des signes d’HTP, le diagnostic clinique semble inadéquat pour la recommandation d’un traitement. Une certitude diagnostique est requise car le traitement adapté est guidé par l’étiologie et la sévérité de l’HTP. Les explorations comprennent une échographie cardiaque et des radiographies du poumon, des tests sanguins pour les maladies associées à l’HTP, la mesure de la capacité à l’effort, de la fonction pulmonaire et des paramètres hémodynamiques par cathétérisme cardiaque. Elles sont, en général, faites en urgence [30].
Classer les patients selon la classification clinique d’HTP est cruciale pour adapter le traitement à la sévérité de la maladie. En particulier, la sécurité et l’efficacité des thérapies pour l’HTP n’ont pas encore été bien établies pour l’hypertension pulmonaire liée à une cardiopathie gauche ou une pathologie pulmonaire avec ou sans hypoxémie. Le diagnostic d’HTP demeure un diagnostic d’exclusion.
Le modèle qui consistait à adresser les patients à des centres spécialisés dans l’HTP a évolué au cours de ces 5 dernières années, le nombre de patients avec hypertension pulmonaire « limite » et insuffisance cardiaque avec fraction d’éjection conservée ayant augmenté. L’échocardiographie seule n’est pas suffisamment sensible pour faire le diagnostic d’HTP et tous les patients dont le diagnostic est douteux doivent avoir un cathétérisme cardiaque afin de mesurer avec précision les paramètres hémodynamiques. Cet examen invasif est souvent utile lorsqu’une pathologie ventriculaire diastolique gauche est suspectée du fait de la coexistence d’hypertension, fibrillation atriale, mais seulement lorsque l’ECG ne permet pas de la mettre en évidence.
De nouveaux outils diagnostiques et de nouvelles approches pour développer ces techniques font l’objet de nombreuses recherches. Elles incluent les techniques écho cardiographiques comme l’imagerie en 3D du ventricule droit. L’IRM est utilisée pour mesurer le volume d’éjection, le volume télédiastolique des ventricules gauche et droit qui peuvent avoir des valeurs pronostiques significatives. Par ailleurs, on a montré que les biomarqueurs sériques comme le NT-proBNP ont une bonne valeur pronostique pour le suivi, bien qu’ils ne soient pas utiles pour le diagnostic positif de l’HTTP.
Alors que le test de marche de 6 min est simple à réaliser pour la plupart des patients, d’autres informations physiopathologiques, comme le mécanisme de la dyspnée, peuvent être obtenues par des tests d’effort cardio-pulmonaires. On sait que la détermination de la consommation maximale d’O2 (VO2 max) a une valeur pronostique, mais des informations supplémentaires peuvent être obtenues à partir de la pente de ventilation/production de CO2, l’oxymètre de pouls et autres mesures de routine.
CONCLUSION
L’HTAP est une maladie rare avec un pronostic sombre. La mortalité de l’HTAP reste élevée avec environ 15% de décès par un an malgré un traitement adapté. Les dépistages et diagnostics précoces de la maladie sont nécessaires avant de procéder au cathétérisme cardiaque droit dans un centre de référence pour le diagnostic de certitude. Etant donné la nature complexe de la pathogénie de l’HTP, la prise en charge multidisciplinaire reste indispensable.
Remerciements
Les auteurs remercient très vivement Madame le Pr F. Capron, Service d’Anatomie et de Cytologie Pathologique - Hôpital Pitié Salpétrière, pour les photographies illustrées dans cet article.
RÉFÉRENCES
1. Sitbon O, Humbert M, Nunes H, Parent F, Garcia G, Hervé P, Rainisio M, Simonneau G. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol 2002 ; 40 : 780-8.
2. Galiè N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J 2009 ; 30 : 2493-537.
3. Galiè N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J 2009 ; 34 : 1219-63.
4. Sommer N, Dietrich A, Schermuly RT, Ghofrani HA, Gudermann T, et al. Regulation of hypoxic pulmonary vasoconstriction: basic mechanisms. Eur Respir J 2008 ; 32 : 1639-51.
5. Gan CT, Lankhaar JW, Westerhof N, Marcus JT, Becker A, Twisk JW, Boonstra A, Postmus PE, Vonk-Noordegraaf A. Noninvasively assessed pulmonary artery stiffness predicts mortality in pulmonary arterial hypertension. Chest 2007 ; 132 : 1906-12.
6. Montani D, Price LC, Dorfmuller P, Achouh L, Jaïs X, Yaïci A, Sitbon O, Musset D, Simonneau G, Humbert M. Pulmonary veno-occlusive disease. Eur Respir J 2009 ; 33 : 189-200.
7. Ito K, Ichiki T, Ohi K, Egashira K, Ohta M, Taguchi K, Takeshita A. Pulmonary capillary hemangiomatosis with severe pulmonary hypertension. Circ J 2003 ; 67 : 793-5.
8. Stewart DJ, Levy RD, Cernacek P, Langleben D. Increased plasma endothelin-1 in pulmonary hypertension: marker or mediator of disease? Ann Intern Med 1991 ; 114 : 464-9.
9. Galie N, Grigioni F, Bacchi-Reggiani L. Relation of endothelin-1 to survival in patients with primary pulmonary hypertension. Eur J Clin Invest 1996 ; 26 (suppl. 1) : 273.
10. Eddahibi S, Humbert M, Fadel E, Raffestin B, Darmon M, Capron F, Simonneau G, Dartevelle P, Hamon M, Adnot S. Serotonin transporter overexpression is responsible for pulmonary artery smooth muscle hyperplasia in primary pulmonary hypertension. J Clin Invest 2001 ; 108 : 1141–50.
11. Izikki M, Hanoun N, Marcos E, Savale L, et al. Tryptophan hydroxylase 1 (Tph1) knock-out and Tph2 polymorphism: effects on hypoxic pulmonary hypertension in mice. A m J Physiol Lung Cell Mol Physiol 2007 ; 293 : L1045–L1052.
12. Rothman RB, Ayestas MA, Dersch CM, Baumann MH. Aminorex, fenfluramine, and chlorphentermine are serotonin transporter substrates-Implications for primary pulmonary hypertension. Circulation 1999 : 100 : 869–75.
13. Keil A, Blom IE, Goldschmeding R, Rupprecht HD. Nitric oxide down-regulates connective tissue growth factor in rat mesangial cells. Kidney Int 2002 ; 62 : 401-11.
14. Emerson M, Momi S, Paul W, Alberti PF, Page C, Gresele P. Endogenous nitric oxide acts as a natural antithrombotic agent in vivo by inhibiting plateletaggregation in the pulmonary vasculature. Thromb Haemost 1999 ; 81 : 961-6.
15. Tahara N, Kai H, Niivama H, et al. Repeated gene transfer of naked prostacyclin synthase plasmid into skeletal muscles attenuates monocrotaline-induced pulmonary hypertension and prolongs survival in rats. Hum Gene Ther 2004;15:1270-8.
16. Tuder RM, Cool CD, Geraci MW, Wang J, Abman SH, Wright L, Badesch D, Voelkel NF. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med 1999 ; 159 : 1925-32.
17. Christman BW, McPherson CD, Newman JH, King GA, Bernard GR, Groves BM, Loyd JE. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med 1992 ; 327 : 70-5.
18. Katugampola SD, Davenport AP. Thromboxane receptor density is increased in human cardiovascular disease with evidence for inhibition at therapeutic concentrations by the AT(1) receptor antagonist losartan. Br J Pharmacol 2001 ; 134:138592.
19. Petkov V, Mosgoeller W, Ziesche R, Raderer M, et al. Vasoactive intestinal peptide as a new drug for treatment of primary pulmonary hypertension. J Clin Invest 2003 ; 111: 1339-46.
20. Said SI, Hamidi SA, Dickman KG, et al. Moderate pulmonary arterial hypertension in male mice lacking the vasoactive intestinal peptide gene. Circulation 2 007 ; 115 : 1260-8.
21. Kovacs G, Berghold A, Scheidl S, Olschewski H. Pulmonary arterial pressure during rest and exercise in healthy subjects: a systematic review. Eur Respir J 2009 ; 34 : 888-94.
22. Montani D, Price LC, Dorfmuller P, Achouh L, et al. Pulmonary veno-occlusive disease. Eur Respir J 2009 ; 33 : 189-200.
23. Dartevelle P, Fadel E, Mussot S, et al. Chronic thromboembolic pulmonary hypertension. Eur Respir J 2004 ; 23 : 637-48.
24. Duong-Quy S. Hypertension artérielle pulmonaire. R e v M al Respir Actual 2010 ; 2 : 27-37.
25. Zlupko M, Harhay MO, Gallop R, Shin J, Archer-Chicko C, Patel R, Palevsky HI, Taichman DB. Evaluation of disease-specific health-related quality of life in patients with pulmonary arterial hypertension. Respir Med 2008 ; 102 : 1431-8.
26. Nagaya N, Uematsu M, Satoh T, Kyotani S, Sakamaki F, Nakanishi N, et al. Serum uric acid levels correlate with the severity and the mortality of primary pulmonary hypertension. Am J Respir Crit Care Med 1999 ; 160 : 487-92.
27. Fijalkowska A, Kurzyna M, Torbicki A, Szewczyk G, Florczyk M, Pruszczyk P, et al. Serum N-terminal brain natriuretic peptide as a prognostic parameter in patients with pulmonary hypertension. Chest 2006 ; 129 : 1313-21.
28. Nagaya N, Nishikimi T, Uematsu M, et al. Plasma brain natriuretic peptide as a prognostic indicator in patients with primary pulmonary hypertension. Circulation 2000; 102 : 865-70.
29. Williams MH, Handler CE, Akram R, Smith CJ, Das C, Smee J, et al. Role of N-terminal brain natriuretic peptide (N-TproBNP) in scleroderma-associated pulmonary arterial hypertension. Eur Heart J 2006 ; 27 : 1485-94.
30. Badesch DB, Champion HC, Sanchez MA, Hoeper MM, Loyd JE, Manes A, et al. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol 2009 ; 54 : S55-66.

FIGURES - TABLES
RÉFÉRENCES
1. Sitbon O, Humbert M, Nunes H, Parent F, Garcia G, Hervé P, Rainisio M, Simonneau G. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol 2002 ; 40 : 780-8.
2. Galiè N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J 2009 ; 30 : 2493-537.
3. Galiè N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J 2009 ; 34 : 1219-63.
4. Sommer N, Dietrich A, Schermuly RT, Ghofrani HA, Gudermann T, et al. Regulation of hypoxic pulmonary vasoconstriction: basic mechanisms. Eur Respir J 2008 ; 32 : 1639-51.
5. Gan CT, Lankhaar JW, Westerhof N, Marcus JT, Becker A, Twisk JW, Boonstra A, Postmus PE, Vonk-Noordegraaf A. Noninvasively assessed pulmonary artery stiffness predicts mortality in pulmonary arterial hypertension. Chest 2007 ; 132 : 1906-12.
6. Montani D, Price LC, Dorfmuller P, Achouh L, Jaïs X, Yaïci A, Sitbon O, Musset D, Simonneau G, Humbert M. Pulmonary veno-occlusive disease. Eur Respir J 2009 ; 33 : 189-200.
7. Ito K, Ichiki T, Ohi K, Egashira K, Ohta M, Taguchi K, Takeshita A. Pulmonary capillary hemangiomatosis with severe pulmonary hypertension. Circ J 2003 ; 67 : 793-5.
8. Stewart DJ, Levy RD, Cernacek P, Langleben D. Increased plasma endothelin-1 in pulmonary hypertension: marker or mediator of disease? Ann Intern Med 1991 ; 114 : 464-9.
9. Galie N, Grigioni F, Bacchi-Reggiani L. Relation of endothelin-1 to survival in patients with primary pulmonary hypertension. Eur J Clin Invest 1996 ; 26 (suppl. 1) : 273.
10. Eddahibi S, Humbert M, Fadel E, Raffestin B, Darmon M, Capron F, Simonneau G, Dartevelle P, Hamon M, Adnot S. Serotonin transporter overexpression is responsible for pulmonary artery smooth muscle hyperplasia in primary pulmonary hypertension. J Clin Invest 2001 ; 108 : 1141–50.
11. Izikki M, Hanoun N, Marcos E, Savale L, et al. Tryptophan hydroxylase 1 (Tph1) knock-out and Tph2 polymorphism: effects on hypoxic pulmonary hypertension in mice. A m J Physiol Lung Cell Mol Physiol 2007 ; 293 : L1045–L1052.
12. Rothman RB, Ayestas MA, Dersch CM, Baumann MH. Aminorex, fenfluramine, and chlorphentermine are serotonin transporter substrates-Implications for primary pulmonary hypertension. Circulation 1999 : 100 : 869–75.
13. Keil A, Blom IE, Goldschmeding R, Rupprecht HD. Nitric oxide down-regulates connective tissue growth factor in rat mesangial cells. Kidney Int 2002 ; 62 : 401-11.
14. Emerson M, Momi S, Paul W, Alberti PF, Page C, Gresele P. Endogenous nitric oxide acts as a natural antithrombotic agent in vivo by inhibiting plateletaggregation in the pulmonary vasculature. Thromb Haemost 1999 ; 81 : 961-6.
15. Tahara N, Kai H, Niivama H, et al. Repeated gene transfer of naked prostacyclin synthase plasmid into skeletal muscles attenuates monocrotaline-induced pulmonary hypertension and prolongs survival in rats. Hum Gene Ther 2004;15:1270-8.
16. Tuder RM, Cool CD, Geraci MW, Wang J, Abman SH, Wright L, Badesch D, Voelkel NF. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med 1999 ; 159 : 1925-32.
17. Christman BW, McPherson CD, Newman JH, King GA, Bernard GR, Groves BM, Loyd JE. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med 1992 ; 327 : 70-5.
18. Katugampola SD, Davenport AP. Thromboxane receptor density is increased in human cardiovascular disease with evidence for inhibition at therapeutic concentrations by the AT(1) receptor antagonist losartan. Br J Pharmacol 2001 ; 134:138592.
19. Petkov V, Mosgoeller W, Ziesche R, Raderer M, et al. Vasoactive intestinal peptide as a new drug for treatment of primary pulmonary hypertension. J Clin Invest 2003 ; 111: 1339-46.
20. Said SI, Hamidi SA, Dickman KG, et al. Moderate pulmonary arterial hypertension in male mice lacking the vasoactive intestinal peptide gene. Circulation 2 007 ; 115 : 1260-8.
21. Kovacs G, Berghold A, Scheidl S, Olschewski H. Pulmonary arterial pressure during rest and exercise in healthy subjects: a systematic review. Eur Respir J 2009 ; 34 : 888-94.
22. Montani D, Price LC, Dorfmuller P, Achouh L, et al. Pulmonary veno-occlusive disease. Eur Respir J 2009 ; 33 : 189-200.
23. Dartevelle P, Fadel E, Mussot S, et al. Chronic thromboembolic pulmonary hypertension. Eur Respir J 2004 ; 23 : 637-48.
24. Duong-Quy S. Hypertension artérielle pulmonaire. R e v M al Respir Actual 2010 ; 2 : 27-37.
25. Zlupko M, Harhay MO, Gallop R, Shin J, Archer-Chicko C, Patel R, Palevsky HI, Taichman DB. Evaluation of disease-specific health-related quality of life in patients with pulmonary arterial hypertension. Respir Med 2008 ; 102 : 1431-8.
26. Nagaya N, Uematsu M, Satoh T, Kyotani S, Sakamaki F, Nakanishi N, et al. Serum uric acid levels correlate with the severity and the mortality of primary pulmonary hypertension. Am J Respir Crit Care Med 1999 ; 160 : 487-92.
27. Fijalkowska A, Kurzyna M, Torbicki A, Szewczyk G, Florczyk M, Pruszczyk P, et al. Serum N-terminal brain natriuretic peptide as a prognostic parameter in patients with pulmonary hypertension. Chest 2006 ; 129 : 1313-21.
28. Nagaya N, Nishikimi T, Uematsu M, et al. Plasma brain natriuretic peptide as a prognostic indicator in patients with primary pulmonary hypertension. Circulation 2000; 102 : 865-70.
29. Williams MH, Handler CE, Akram R, Smith CJ, Das C, Smee J, et al. Role of N-terminal brain natriuretic peptide (N-TproBNP) in scleroderma-associated pulmonary arterial hypertension. Eur Heart J 2006 ; 27 : 1485-94.
30. Badesch DB, Champion HC, Sanchez MA, Hoeper MM, Loyd JE, Manes A, et al. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol 2009 ; 54 : S55-66.
ARTICLE INFO
DOI: 10.12699/jfvp.1.1.2010.35
Conflict of Interest
Non
Date of manuscript receiving
24/4/2010
Date of publication after correction
15/8/2010
Article citation
Duong-Quy S, Le-Dong N.N, Duong-Ngo C, Mai Huu Thanh B, Hua-Huy T, Dinh-Xuan A.T. New definition and classification of Pulmonary Hypertension. J Func Vent Pulm 2010;01(01):35-41.



Copyright: jfvpulm.com