
 English
English
 French
French
 Pubmed
Pubmed Google Scholar
Google Scholar Cross Ref
Cross Ref Visitor 725689
Visitor 725689Rare cause of fatal dyspnea during systemic lupus erythematosus: Pulmonary arterial hypertension
Une cause rare de dyspnée fatale au cours du lupus érythémateux disséminé: L’hypertension artérielle pulmonaire
S. Ksir, A. El Aissaoui, N. Naji, S.El Khader, M. El Aissate, A. Zinebi, MK.Moudden
Service de Médecine Interne
Hôpital militaire Moulay Ismail, BP S15, Meknès, Maroc
Corresponding author
Dr. Ksir Salma
Hospital Militaire Moulay Ismail. Meknes, Maroc
E-mail: Salma.ksir@gmail.com
ABSTRACT
Introduction. Pulmonary arterial hypertension is a serious complication of systemic lupus erythematosus. Its occurrence can be fatal especially in the absence of early treatment.
Case report. We reported a 50 years old patient who had a well-controlled systemic lupus erythematosus. She presented dyspnea NYHA III. Investigations found out the pulmonary arterial hypertension was about 50mmHg. Despite well conducted conventional treatment (corticosteroids and cyclophosphamides) but untaken pulmonary vasodilators, the evolution was fatal.
Conclusion. Pulmonary arterial hypertension is a rare cause of dyspnea during systemic lupus erythematosus. Its prognosis is very bad and early treatment with close monitoring should be considered.
KEYWORDS: Pulmonary arterial hypertension; Systemic lupus erythematosus.
Introduction. L’hypertension artérielle pulmonaire est une complication rare du lupus érythémateux systémique. Sa survenue peut être mortelle surtout en absence du traitement précoce.
Observation. Nous rapportons le cas d’une patiente de 50ans, suivie pour lupus érythémateux disséminé bien contrôlé sous corticothérapie et anti paludéen de synthèse. Elle a présenté une dyspnée stade III de la NYHA. Le bilan étiologique a révélé une hypertension artérielle pulmonaire estimée à 50mmHg. Malgré un traitement conventionnel bien conduit (corticothérapie et cyclophosphamides), mais un traitement vasodilatateur artérielle pulmonaire non pris, l’évolution a été rapidement fatale.
Conclusion. La dyspnée au cours du lupus érythémateux disséminé peut rarement révéler une hypertension artérielle pulmonaire. Cette complication est de pronostic très sombre et le traitement précoce avec une surveillance étroite doivent être envisagés.
MOTS CLÉS: Hypertension artérielle pulmonaire; Lupus érythémateuse systémique.
INTRODUCTION
Le lupus érythémateux dissémine (LES) est une pathologie auto-immune et inflammatoire dont le pronostic est en fonction de la nature des lésions viscérales, en particulier les atteintes rénales et vasculaires. L’hypertension artérielle pulmonaire (HTAP) est une complication rare mais grave du LES. L’insuffisance cardiaque droite est la conséquence de la maladie et sa survenue reste l’élément pronostique majeur. Nous rapportons un cas de LES compliquée d’une HTAP aboutissant à une insuffisance cardiaque droite aigue et au décès.
OBSERVATION
Mme R.E était âgée de 50 ans, Admise au service le mois 06/2018 pour bilan étiologique d’une dyspnée stade III de la NYHA. Elle avait comme antécédent familial : une sœur jumelle décédée en 2009 pour insuffisance rénale probablement d’origine systémique, non documentée. Et comme antécédent personnel : Un lupus érythémateux disséminé depuis 2009 retenu sur les critères SLICC devant les manifestations suivantes (cutanéomuqueuses : érythème malaire, photosensibilité, phénomène de raynaud / ostéo articulaires : type arthrite non érosive non déformante / rénale : protéinurie de 24h positive à 819 mg/l/ immunologique : Anticorps anti nucléaires positifs à 1/1600, aspect moucheté, anti DNA natifs positifs, facteur rhumatoïde positif). La patiente a été mise initialement sous corticothérapie orale à des doses variables (60 à 10mg/jour) associé à un traitement adjuvant et un anti paludéen de synthèse. L’évolution était favorable avec une amélioration clinique et biologique.
En avril 2018, la patiente a présenté une dyspnée d’apparition rapidement progressive stade III de la NYHA pour laquelle elle était hospitalisée dans notre service. La patiente rapportait également la notion d’une douleur thoracique rétrostérnale avec une toux non productive. Le tout évoluant dans un contexte d’apyrexie et d’asthénie généralisée.
L’examen clinique trouvait une patiente consciente, apyrétique, une fréquence respiratoire à 23 cycles/min, une fréquence cardiaque à 88 battements/min, une tension artérielle à 15/07 mmHg. L’auscultation cardiaque avait objectivé un souffle holosystolique d’insuffisance tricuspidienne. L’auscultation pulmonaire était normale. Le reste de l’examen somatique était sans particularités.
La patiente avait bénéficié d’une radiographie thoracique ayant objectivé une cardiomégalie sans épanchement pleural, un éléctrocardiogramme en faveur d’une tachycardie sinusale.
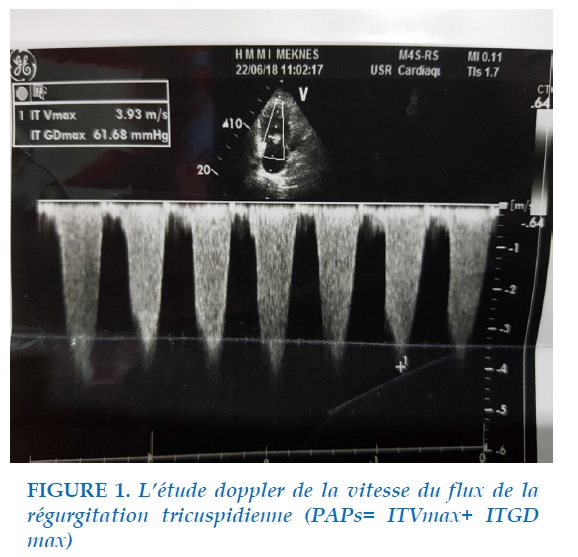
Une échographie transthoracique (Figure 1) revenant en faveur d’un cœur pulmonaire chronique, ventricule droit dilatée, oreillette droite dilatée, hypertension artérielle pulmonaire estimée à 66mmHg, ventricule gauche non dilaté légèrement hypertrophié, cinétique paradoxale du septum, insuffisance tricuspidienne modérée. L’angioscanner thoracique réalisé était sans anomalies. Une tomodensitométrie thoracique objectivant un syndrome interstitiel basal bilatéral fait d’images réticulées diffuses. La patiente était mise sous diurétique de l’anse furosémide 80mg/jour en deux injections, et antivitamines K avec contrôle serré de l’INR. La patiente a bénéficié ensuite d’un cathétérisme droit confirmant une hypertension artérielle pulmonaire pré-capillaire. Il s’agissait donc d’un lupus érythémateux disséminé compliqué d’une HTAP pré-capillaire.
La patiente a bénéficié d’un traitement immunosuppresseur à base de corticothérapie forte dose 1mg/kg/jour et un bolus de cyclophosphamides. Elle a bénéficié également d’une prescription d’un traitement vasodilatateur artérielle pulmonaire à base d’analogues d’endothéline à la dose de 62.5mg deux fois par jour pendant 04 semaines puis une augmentation par palier jusqu’à une posologie d’entretien de 125mg deux fois par jour. Pour des problèmes d’assurance, la patiente n’a pas pu avoir les analogues d’endothéline. 2 mois plus tard, la patiente était admise aux urgences puis en réanimation où elle a succombée, après 01 mois de soins intensifs, à une insuffisance ventriculaire droite aigue sur HTAP.
DISCUSSION
L’Hypertension artérielle pulmonaire est une complication rare et sévère des connectivites, qui concerne avant tout la sclérodermie systémique, mais également le lupus systémique avec une prévalence variant entre 0,5 à 17,5 % [1]. L’HTAP au cours du lupus, peut-être d’allure primitive ou secondaire [2] et survient généralement après plusieurs années d’évolution en moyenne de 7,4 ± 6,5 ans dans la série de Shen et al. [3], et 14 ± 6 ans dans la série de Winslow et al. [4]. Dans notre observation, l’HTAP est probablement d’allure primitive ou secondaire à la pneumopathie infiltrative diffuse lupique objectivée à la TDM. Le délai de la survenue de l’HTAP chez cette patiente suivie de LES était de 8 ans, ce qui rejoint les données de la littérature. D’autres travaux se sont intéressés à étudier le profil des patients lupiques pouvant se compliquer d’une HTAP. En effet, il a été démontré que les patients avec un bilan immunologique positif du facteur rhumatoïde, d’anticorps anti-Sm ou d’anticorps anti-RNP sont les plus à risque [3,5]. L’existence d’une association la survenue d’un phénomène de Raynaud et l’HTAP a été évoquée au cours du LES ce qui signifie qu'il existerait une composante vasospastique pulmonaire chez ces patients mais cette association reste très controversée [6,7]. A relever que l'HTP avec phénomène de Raynaud est associée à une diminution de la survie [8] .Notre patiente avait effectivement un syndrome de raynaud et une recherche positive du facteur rhumatoïde. Pour certains auteurs, la survenue de l’HTP est en relation avec l’activité du LES sous-jacent [9] alors que pour d’autres, non [10]. L’activité clinique et biologique du lupus était contrôlée chez notre patiente.
L’HTAP se définit comme l’ensemble des maladies dont l’origine se situe au niveau des petites artères pulmonaires avec une pression artérielle pulmonaire (PAP) moyenne supérieure à 25 mmHg au repos, mesurée lors du cathétérisme cardiaque droit [1]. L’échocardiographie représente l’examen de choix pour le dépistage initial de l’HTAP. Cette pression était estimée à 66mmHg à l’échocardiographie chez notre patiente et à 50mmHg au cathétérisme cardiaque droit.
Le traitement de l’HTAP au cours du Lupus systémique est fonction des phénomènes physiopathologiques impliqués (origine auto-immun, thrombo-embolique, ou d’allure primitive). Lorsque l’HTP est secondaire à une PID lupique, le traitement associe corticothérapie et immunosuppresseurs. Lorsque l’HTP est d’allure « primitive », le traitement associe des recommandations aux patients, des mesures symptomatiques, un traitement vasodilatateur pulmonaire à partir d’une dyspnée classe fonctionnelle II de la NYHA, et un traitement immunosuppresseur [11]. Les anticoagulants visent à éviter les embolies pulmonaires et les thromboses in situ, facteurs d’aggravation de l’HTAP. Le pronostic d’une HTAP non traitée est mauvais et comparable à celui d’une maladie oncologique avancée [12].
L’originalité de notre observation repose sur la survenue d’une HTAP ayant été rapidement mortelle malgré un traitement immunosuppresseur et anticoagulant bien conduit mais un traitement vasodilatateur artériel pulmonaire non pris.
CONCLUSION
L’hypertension artérielle pulmonaire est une complication relativement rare mais potentiellement mortelle du lupus érythémateux disséminé. La survenue de cette complication a eu lieu chez notre patiente malgré une activité contrôlée de son lupus. Le pronostic était rapidement sombre et le décès a eu lieu suite à une insuffisance ventriculaire droite aigue.
CONFLITS D’INTÉRÊT
Les auteurs ne déclarent aucun conflit d’intérêt.
RÉFÉRENCES
1. L. Arnauda , C. Agardc, J. Haroche, P. Cacoub, J.-C. Piette, Z. Amoura. Hypertension artérielle pulmonaire associée au lupus systémique. La Revue de médecine interne 32 (2011) 689–697.
2. Galie N, Hoeper MM, Humbert M, Torbicki A, Vachi-ery JL, Barbera JA, et al. Guidelines for the diagnosiis and treatment of pulmonary hypertension. Eur Respir J 2009;34:1219–63.
3. Tanaka E, Harigai M, Tanaka M, Kawaguchi Y, Hara M, Kamatani N. Pulmonary hypertension in systemic lupus erythematosus: evaluation of clinical characteris-tics and response to immunosuppressive treatment. J Rheumatol 2002;29:282–7.
4. Winslow TM, Ossipov MA, Fazio GP, Simonson JS, Redberg RF, Schiller NB. Five-year follow-up study of the prevalence and progression of pulmonary hyper-tension in systemic lupus erythematosus. Am Heart J 1995;129: 510–5.
5. Pan TL, Thumboo J, Boey ML. Primary and secondary pulmonary hypertension in systemic lupus erythema-tosus. Lupus 2000;9:338–42.
6. Quismorio Jr FP, Sharma O, Koss M, Boylen T, Edmis-ton AW, Thornton PJ, et al. Immunopathologic and clinical studies in pulmonary hypertension associated with systemic lupus erythematosus. Semin Arthritis Rheum 1984;13:349–59
7. Foïs E. Prévalence de l’hypertension arterielle pulmo-naire dans le lupus erythemateux systemique. Dépis-tage par échocardiographie. [thèse]. Paris: Université Paris 11; 2006.
8. J.-M. Fellrath A. Sauty. Lupus érythémateux et atteinte respiratoire. Rev Med Suisse 2000; volume -4. 20367.
9. Shen JY, Chen SL, Wu YX, Tao RQ, Gu YY, Bao CD, et al. Pulmonary hypertension in systemic lupus erythe-matosus. Rheumatol Int 1999;18:147–51.
10. Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Pulmonary arterial hypertension in france: results from a national registry. Am J Respir Crit Care Med 2006;173:1023–30.
11. Haute autorité de santé. Hypertension artérielle pul-monaire, protocole national de diagnostic et de soins pour une maladie rare. 1997.
12. Andrea Azzola. Hypertension pulmonaire pour le généraliste en 2018. Rev Med Suisse 2018; volume 14. 212-215

FIGURE
RÉFÉRENCES
1. L. Arnauda , C. Agardc, J. Haroche, P. Cacoub, J.-C. Piette, Z. Amoura. Hypertension artérielle pulmonaire associée au lupus systémique. La Revue de médecine interne 32 (2011) 689–697.
2. Galie N, Hoeper MM, Humbert M, Torbicki A, Vachi-ery JL, Barbera JA, et al. Guidelines for the diagnosiis and treatment of pulmonary hypertension. Eur Respir J 2009;34:1219–63.
3. Tanaka E, Harigai M, Tanaka M, Kawaguchi Y, Hara M, Kamatani N. Pulmonary hypertension in systemic lupus erythematosus: evaluation of clinical characteris-tics and response to immunosuppressive treatment. J Rheumatol 2002;29:282–7.
4. Winslow TM, Ossipov MA, Fazio GP, Simonson JS, Redberg RF, Schiller NB. Five-year follow-up study of the prevalence and progression of pulmonary hyper-tension in systemic lupus erythematosus. Am Heart J 1995;129: 510–5.
5. Pan TL, Thumboo J, Boey ML. Primary and secondary pulmonary hypertension in systemic lupus erythema-tosus. Lupus 2000;9:338–42.
6. Quismorio Jr FP, Sharma O, Koss M, Boylen T, Edmis-ton AW, Thornton PJ, et al. Immunopathologic and clinical studies in pulmonary hypertension associated with systemic lupus erythematosus. Semin Arthritis Rheum 1984;13:349–59
7. Foïs E. Prévalence de l’hypertension arterielle pulmo-naire dans le lupus erythemateux systemique. Dépis-tage par échocardiographie. [thèse]. Paris: Université Paris 11; 2006.
8. J.-M. Fellrath A. Sauty. Lupus érythémateux et atteinte respiratoire. Rev Med Suisse 2000; volume -4. 20367.
9. Shen JY, Chen SL, Wu YX, Tao RQ, Gu YY, Bao CD, et al. Pulmonary hypertension in systemic lupus erythe-matosus. Rheumatol Int 1999;18:147–51.
10. Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Pulmonary arterial hypertension in france: results from a national registry. Am J Respir Crit Care Med 2006;173:1023–30.
11. Haute autorité de santé. Hypertension artérielle pul-monaire, protocole national de diagnostic et de soins pour une maladie rare. 1997.
12. Andrea Azzola. Hypertension pulmonaire pour le généraliste en 2018. Rev Med Suisse 2018; volume 14. 212-215
ARTICLE INFO DOI: 10.12699/jfvpulm.11.35.2020.49 Conflict of Interest



Copyright: jfvpulm.com